Marwaha Ashish, Ibrahim Judy, Rice Taylor, Hamwi Nadia, Rupar Charles Anthony, Cresswell David, Prasad Chitra, Schulze Andreas
Clinical and Metabolic Genetics The Hospital for Sick Children Toronto Ontario Canada.
Department of Academic Affairs Tawam Hospital Al Ain Abu Dhabi United Arab Emirates.
JIMD Rep. 2020 Oct 1;57(1):9-14. doi: 10.1002/jmd2.12171. eCollection 2021 Jan.
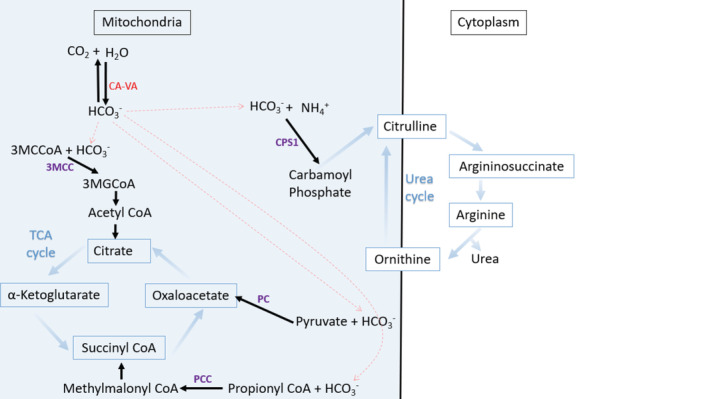
The combination of neonatal hyperammonemia, lactic acidosis, ketonuria, and hypoglycemia is pathognomonic for carbonic anhydrase VA (CA-VA) deficiency. We present two cases of this rare inborn error of metabolism. Both newborns with South Asian ancestry presented with a metabolic decompensation characterized by hyperammonemia, lactic acidosis and ketonuria; one also had hypoglycemia. Standard metabolic investigations (plasma amino acids, acylcarnitine profile, and urine organic acids) were not indicative of a specific organic aciduria or fatty acid oxidation defect but had some overlapping features with a urea cycle disorder (elevated glutamine, orotic acid, and low arginine). Hyperammonemia was treated initially with nitrogen scavenger therapy and carglumic acid. One patient required hemodialysis. Both have had a favorable long-term prognosis after their initial metabolic decompensation. Genetic testing confirmed the diagnosis of carbonic anhydrase VA (CA-VA) deficiency due to biallelic pathogenic variants in . These cases are in line with 15 cases previously described in the literature, making the phenotypic presentation pathognomonic for this ultrarare (potentially underdiagnosed) inborn error of metabolism with a good prognosis.
新生儿高氨血症、乳酸酸中毒、酮尿症和低血糖同时出现是碳酸酐酶VA(CA-VA)缺乏症的特征性表现。我们报告了两例这种罕见的先天性代谢缺陷病例。两名具有南亚血统的新生儿均出现以高氨血症、乳酸酸中毒和酮尿症为特征的代谢失代偿;其中一名还伴有低血糖。标准代谢检查(血浆氨基酸、酰基肉碱谱和尿有机酸)未提示特定的有机酸尿症或脂肪酸氧化缺陷,但具有一些与尿素循环障碍重叠的特征(谷氨酰胺、乳清酸升高,精氨酸降低)。高氨血症最初采用氮清除疗法和卡谷氨酸治疗。一名患者需要进行血液透析。两名患者在最初的代谢失代偿后均有良好的长期预后。基因检测证实诊断为碳酸酐酶VA(CA-VA)缺乏症,原因是存在双等位基因致病性变异。这些病例与文献中先前描述的15例病例一致,使得这种超罕见(可能诊断不足)的先天性代谢缺陷的表型表现具有特征性,且预后良好。