Division of Pulmonology, Department of Internal Medicine, College of Medicine, Chungnam National University, Daejeon, Republic of Korea.
Division of Chemical and Biological metrology, Korea Research Institute for Standards and Science, Daejeon, South Korea.
Cancer Med. 2021 Feb;10(4):1405-1417. doi: 10.1002/cam4.3734. Epub 2021 Jan 23.
Despite the progress of advanced target therapeutic agents and immune checkpoint inhibitors, EGFR-TKI resistance is still one of the biggest obstacles in treating lung cancer. Clinical studies with autophagy inhibitors are actively underway to overcome drug resistance.
We used PC9, PC9/GR, and HCC827/GR cell lines to evaluate the activation of autophagy and EGFR-TKI resistance. Chloroquine was applied as an autophagic blocker and verteporfin was utilized as a YAP inhibitor.
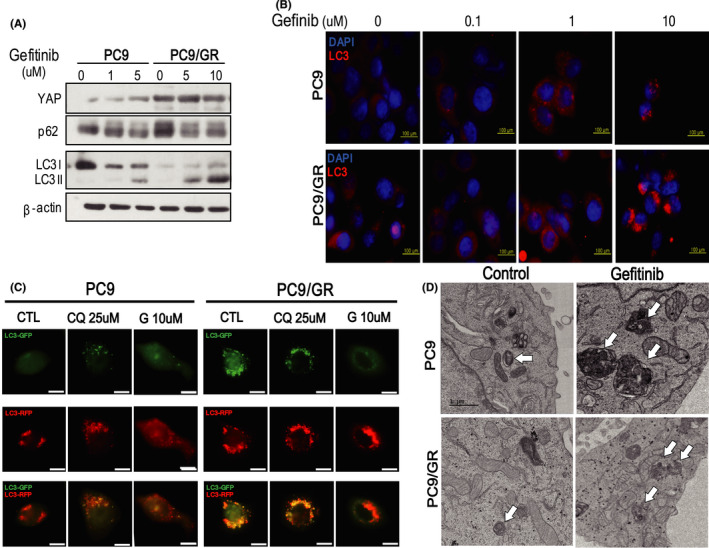
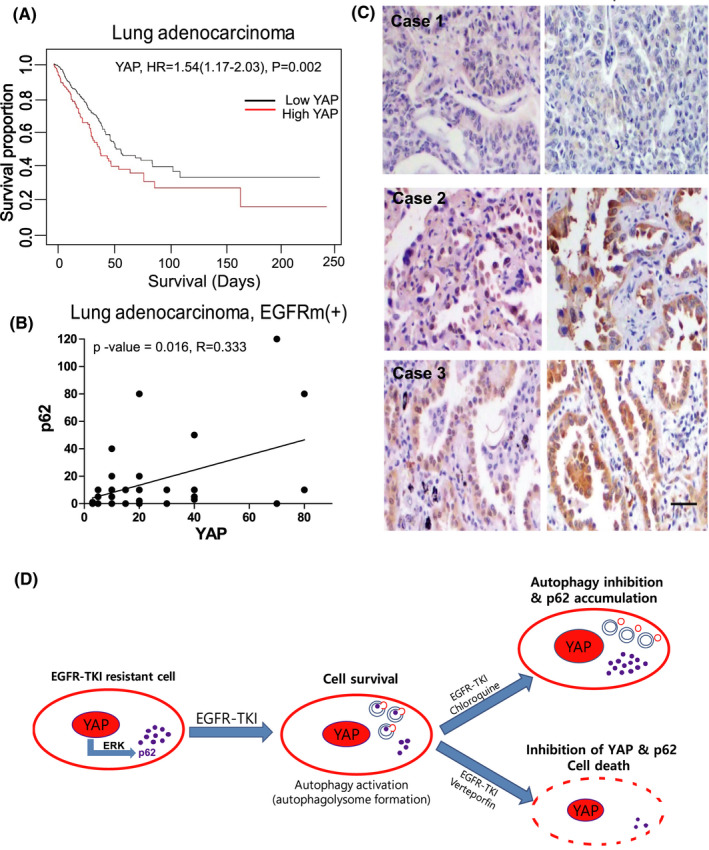
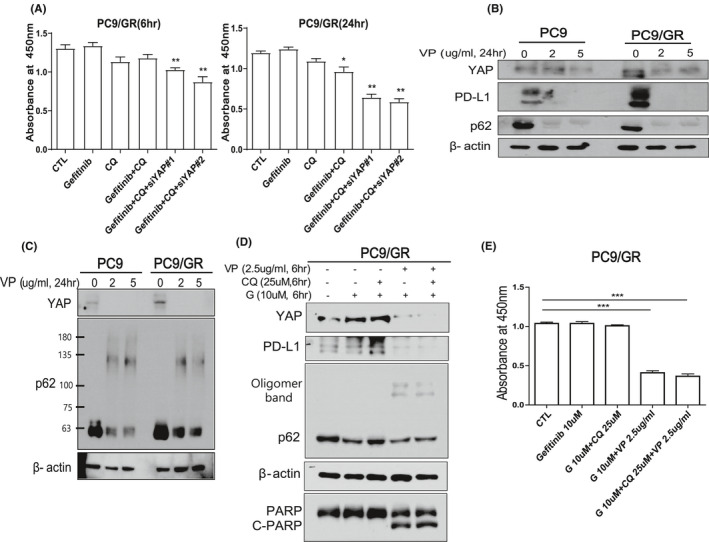
In this study, we tried to reveal the effect of autophagy adaptor p62 which is accumulated by autophagy inhibitor in EGFR-TKI-resistant lung adenocarcinoma. We identified that p62 has oncogenic functions that induce cell proliferation and invasion of EGFR-TKI-resistant lung adenocarcinoma. Interestingly, we found for the first time that YAP regulates p62 transcription through ERK, and YAP inhibition can suppress the expression of oncogenic p62. We also confirmed that the expressions of p62 and YAP have a positive correlation in EGFR-mutant lung adenocarcinoma patients. To block cell survival via perturbing YAP-p62 axis, we treated EGFR-TKI-resistant lung cancer cells with YAP inhibitor verteporfin. Remarkably, verteporfin effectively caused the death of EGFR-TKI-resistant lung cancer cells by decreasing the expressions of p62 with oncogenic function, YAP, and its target PD-L1. So, the cumulative effect of oncogenic p62 should be considered when using autophagy inhibitors, especially drugs that act at the last stage of autophagy such as chloroquine and bafilomycin A1.
Finally, we suggest that targeting YAP-p62 signaling axis can be useful to suppress the EGFR-TKI-resistant lung cancer. Therefore, drug repurposing of verteporfin for lung cancer treatment may be valuable to consider because it can inhibit critical targets: p62, YAP, and PD-L1 at the same time.
尽管先进的靶向治疗药物和免疫检查点抑制剂取得了进展,但 EGFR-TKI 耐药仍然是治疗肺癌的最大障碍之一。目前正在积极开展使用自噬抑制剂的临床研究,以克服耐药性。
我们使用 PC9、PC9/GR 和 HCC827/GR 细胞系来评估自噬的激活和 EGFR-TKI 耐药性。氯喹被用作自噬阻断剂,而维替泊芬被用作 YAP 抑制剂。
在这项研究中,我们试图揭示自噬接头蛋白 p62 在 EGFR-TKI 耐药肺腺癌中的作用,该蛋白是由自噬抑制剂积累的。我们发现 p62 具有致癌功能,可诱导 EGFR-TKI 耐药肺腺癌的细胞增殖和侵袭。有趣的是,我们首次发现 YAP 通过 ERK 调节 p62 的转录,并且 YAP 抑制可以抑制致癌 p62 的表达。我们还证实,在 EGFR 突变型肺腺癌患者中,p62 和 YAP 的表达呈正相关。为了通过扰乱 YAP-p62 轴来阻断细胞存活,我们用 YAP 抑制剂维替泊芬处理 EGFR-TKI 耐药的肺癌细胞。值得注意的是,维替泊芬通过降低具有致癌功能的 p62、YAP 及其靶标 PD-L1 的表达,有效地导致 EGFR-TKI 耐药的肺癌细胞死亡。因此,在使用自噬抑制剂时,应考虑到致癌性 p62 的累积效应,特别是作用于自噬最后阶段的药物,如氯喹和巴弗洛霉素 A1。
最后,我们建议靶向 YAP-p62 信号通路可能有助于抑制 EGFR-TKI 耐药的肺癌。因此,将维替泊芬用于肺癌治疗的药物再利用可能是值得考虑的,因为它可以同时抑制关键靶点:p62、YAP 和 PD-L1。