Alqanatish Jubran, Alsowailmi Banan, Alfarhan Haneen, Alhamzah Albandari, Alharbi Talal
King Saud Bin Abdulaziz University for Health Sciences (KSAU-HS), Riyadh 14611, Saudi Arabia.
King Abdullah International Medical Research Center (KAIMRC), Riyadh 14611, Saudi Arabia.
Open Access Rheumatol. 2021 Jan 15;13:15-21. doi: 10.2147/OARRR.S276112. eCollection 2021.
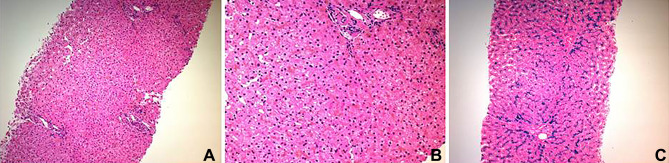
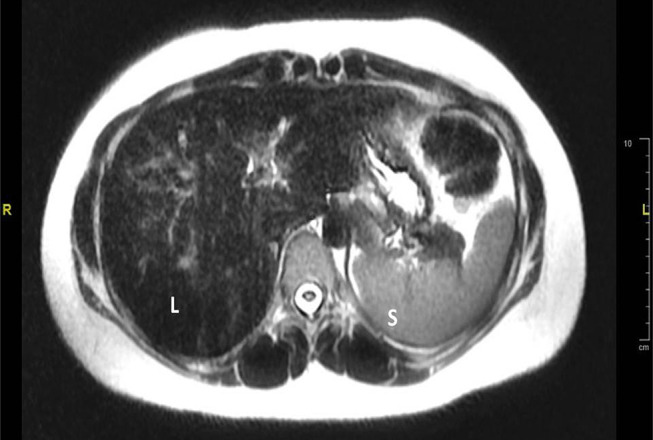
Hereditary hemochromatosis (HH) is an inherited iron overload. The most common form of HH is type 1 hereditary hemochromatosis (HFE-related), which is associated with mutation of the HFE gene located on chromosome 6 and inherited in an autosomal recessive pattern. Type 2 hereditary hemochromatosis or juvenile hemochromatosis is less frequent autosomal recessive disease that results from mutations in the HJV gene on chromosome 1 (type2a) or the HAMP gene on chromosome19 (type2b). Mutation of type 2 transferrin receptor gene and mutation of the ferroportin gene result in hemochromatosis type 3 and hemochromatosis type 4, respectively. Juvenile hemochromatosis is characterized by an early onset of excess accumulation of iron in various organs. It could affect the liver, heart, pancreas and joints, resulting in arthropathy. Most juvenile hemochromatosis cases exhibit severe symptoms due to early onset. Cardiac and hypogonadism are the dominating features of the disease. Prevalence of arthropathy in juvenile hemochromatosis is higher than classic HH. Early diagnosis and intervention of juvenile hemochromatosis may prevent irreversible organ damage. The diagnosis can be made based on laboratory testing (of increased transferrin saturation, serum iron and ferritin levels), liver biopsy, imaging or genotype. According to international guidelines, treatment of HH is indicated when serum ferritin concentrations are above the upper limit of normal. We report two sisters who presented to the rheumatology clinic with arthralgia, which was subsequently found to have a homozygous mutation variant of unknown significance in the HFE2 gene: c.497A>G;p.(His166Arg) and has been treated with deferasirox (Exjade®). Musculoskeletal symptoms completely resolved in both patients in two months and remained so for one year on treatment.
遗传性血色素沉着症(HH)是一种遗传性铁过载疾病。HH最常见的形式是1型遗传性血色素沉着症(与HFE相关),它与位于6号染色体上的HFE基因突变有关,并以常染色体隐性模式遗传。2型遗传性血色素沉着症或青少年血色素沉着症是一种较罕见的常染色体隐性疾病,由1号染色体上的HJV基因(2a型)或19号染色体上的HAMP基因(2b型)突变引起。2型转铁蛋白受体基因突变和铁转运蛋白基因突变分别导致3型血色素沉着症和4型血色素沉着症。青少年血色素沉着症的特征是在各个器官中过早出现铁过量积累。它可能影响肝脏、心脏、胰腺和关节,导致关节病。大多数青少年血色素沉着症病例由于发病早而表现出严重症状。心脏和性腺功能减退是该疾病的主要特征。青少年血色素沉着症中关节病的患病率高于经典HH。青少年血色素沉着症的早期诊断和干预可能预防不可逆的器官损伤。诊断可基于实验室检测(转铁蛋白饱和度、血清铁和铁蛋白水平升高)、肝活检、影像学检查或基因分型。根据国际指南,当血清铁蛋白浓度高于正常上限时,需对HH进行治疗。我们报告了两名姐妹,她们因关节痛到风湿病诊所就诊,随后发现其HFE2基因存在意义不明的纯合突变变体:c.497A>G;p.(His166Arg),并接受了地拉罗司(Exjade®)治疗。两名患者的肌肉骨骼症状在两个月内完全缓解,治疗一年后仍保持缓解状态。