Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN, USA.
Institut für Medizinische Mikrobiologie, Universität Zürich, Zürich, Switzerland.
J Antibiot (Tokyo). 2021 Jun;74(6):381-396. doi: 10.1038/s41429-021-00408-3. Epub 2021 Jan 27.
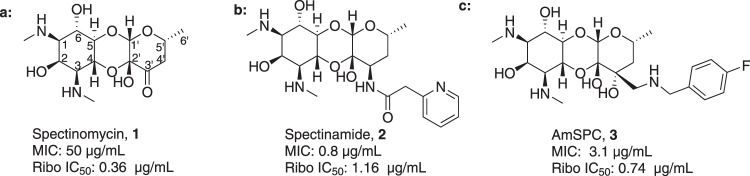
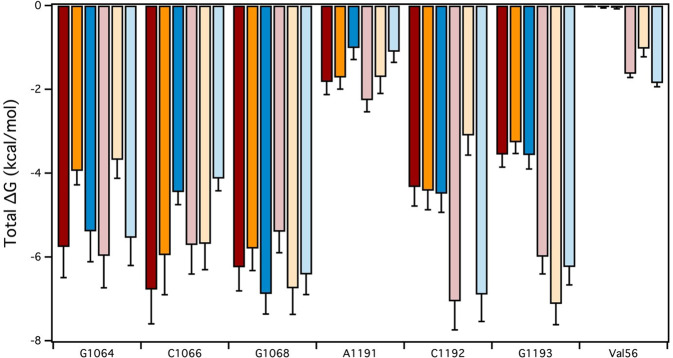
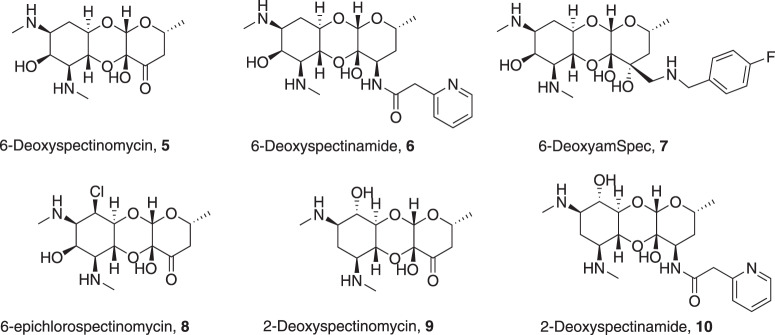
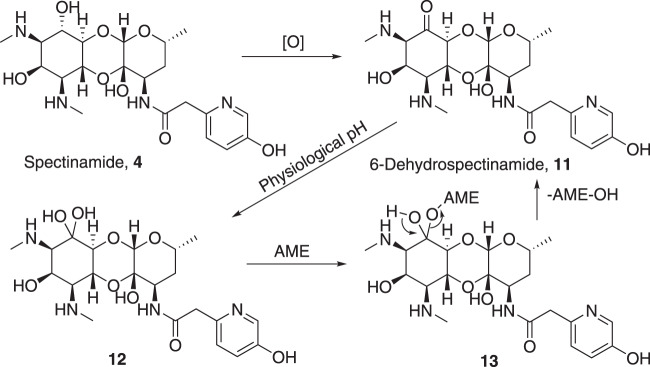
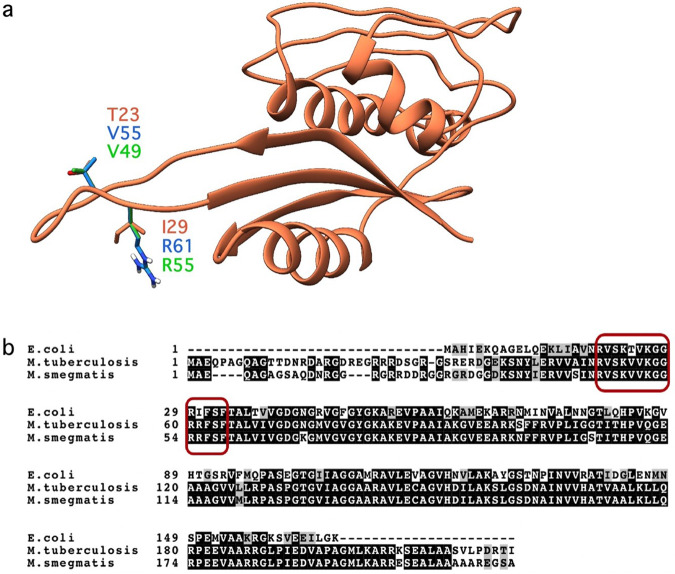
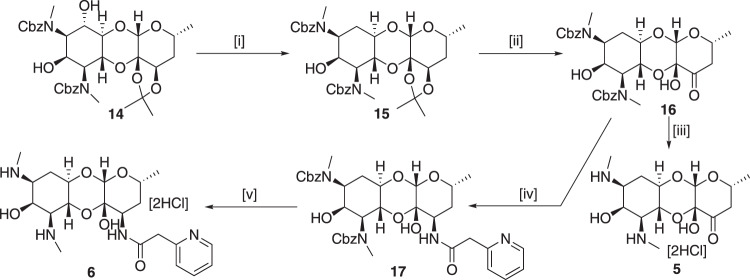
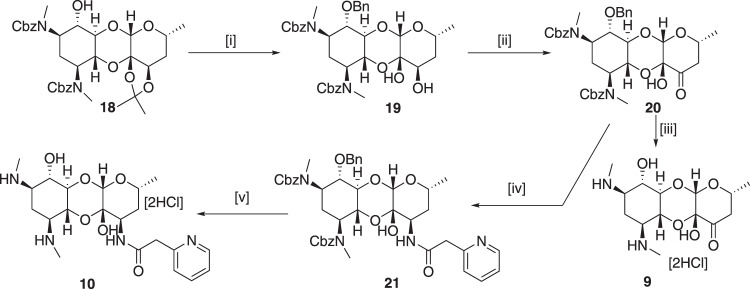
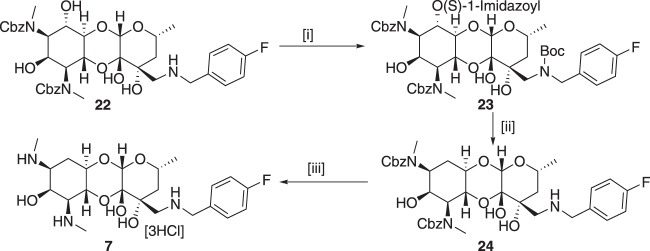
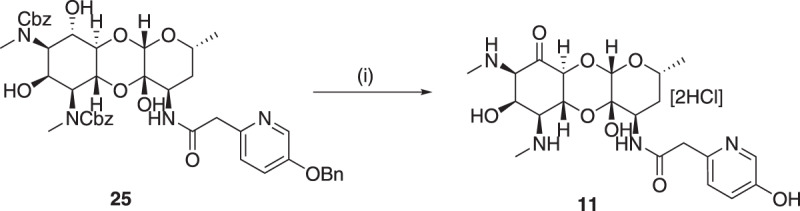
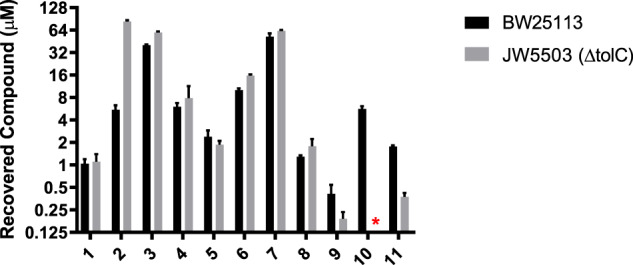
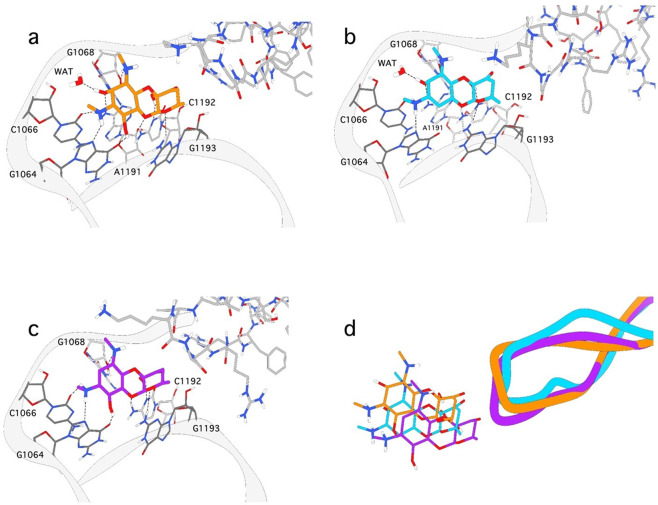
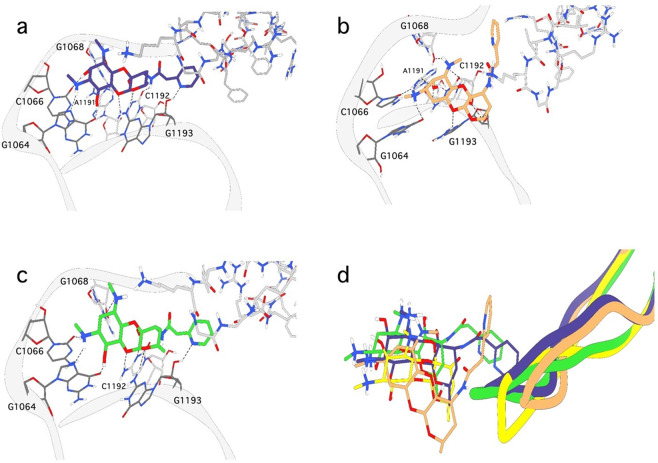
Spectinomycin, an aminocyclitol antibiotic, is subject to inactivation by aminoglycoside modifying enzymes (AMEs) through adenylylation or phosphorylation of the 6-hydroxy group position. In this study, the effects of deoxygenation of the 2- and 6-hydroxy group positions on the spectinomycin actinamine ring are probed to evaluate their relationship to ribosomal binding and the antimicrobial activities of spectinomycin, semisynthetic aminomethyl spectinomycins (amSPCs), and spectinamides. To generate these analogs, an improved synthesis of 6-deoxyspectinomycin was developed using the Barton deoxygenation reaction. 6-Dehydrospectinamide was also synthesized from spectinamide 4 to evaluate the H-bond acceptor character on the C-6 position. All the synthesized analogs were tested for antibacterial activity against a panel of Gram (+) and Gram (-) pathogens, plus Mycobacterium tuberculosis. The molecular contribution of the 2- and 6-hydroxy group and the aryl functionalities of all analogs were examined by measuring inhibition of ribosomal translation and molecular dynamics experiments with MM/GBSA analysis. The results of this work indicate that the 6-hydroxy group, which is the primary target of AMEs, is a required motif for antimicrobial activity in current analogs. Removal of the 6-hydroxy group could be partially rescued by offsetting ribosomal binding contributions made by the aryl side chains found in the spectinamide and amSPCs. This study builds on the knowledge of the structure-activity relationships of spectinomycin analogs and is being used to aid the design of next-generation spectinomycins.
壮观霉素是一种氨基环醇类抗生素,可通过氨基糖苷修饰酶(AMEs)使 6-羟基发生腺苷酰化或磷酸化而失活。在这项研究中,研究了 2-位和 6-位羟基脱氧对壮观霉素氨甲酰基环的影响,以评估它们与核糖体结合的关系以及壮观霉素、半合成氨甲基壮观霉素(amSPCs)和壮观霉素酰胺的抗菌活性。为了生成这些类似物,使用 Barton 脱氧反应开发了一种改进的 6-去氧壮观霉素合成方法。还从壮观霉素 4 合成了 6-脱水壮观霉素酰胺,以评估 C-6 位的氢键供体特性。所有合成的类似物均针对一组革兰氏阳性和革兰氏阴性病原体以及结核分枝杆菌进行了抗菌活性测试。通过测量核糖体翻译抑制和 MM/GBSA 分析的分子动力学实验,研究了所有类似物的 2-和 6-羟基基团以及芳基官能团的分子贡献。这项工作的结果表明,6-羟基基团是 AMEs 的主要靶标,是当前类似物中抗菌活性所必需的基序。通过抵消在壮观霉素酰胺和 amSPCs 中发现的芳基侧链对核糖体结合的贡献,可以部分挽救去除 6-羟基基团的影响。本研究建立在壮观霉素类似物的结构-活性关系的知识基础上,并正在用于辅助下一代壮观霉素的设计。