Ammer Luise Sophie, Pohl Sandra, Breyer Sandra Rafaela, Aries Charlotte, Denecke Jonas, Perez Anna, Petzoldt Martin, Schrum Johanna, Müller Ingo, Muschol Nicole Maria
Department of Pediatrics, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
International Center for Lysosomal Disorders (ICLD), University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Mol Genet Metab Rep. 2021 Jan 14;26:100704. doi: 10.1016/j.ymgmr.2020.100704. eCollection 2021 Mar.
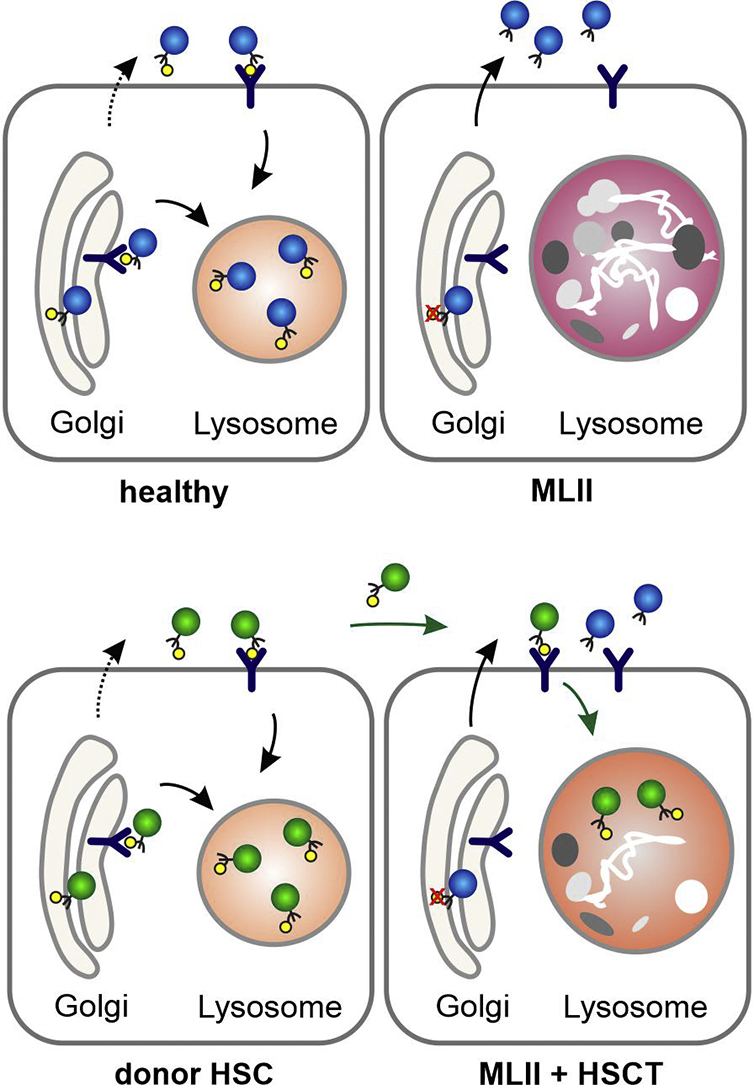
Mucolipidosis type II (MLII) is an ultra-rare lysosomal storage disorder caused by defective lysosomal enzyme trafficking. Clinical hallmarks are craniofacial dysmorphia, cardiorespiratory dysfunction, hepatosplenomegaly, skeletal deformities and neurocognitive retardation. Death usually occurs in the first decade of life and no cure exists. Hematopoietic stem cell transplantation (HSCT) has been performed in few MLII patients, but comprehensive follow-up data are extremely scarce.
MLII diagnosis was confirmed in a female three-month-old patient with the mutations c.2213C > A and c.2220_2221dup in the gene. At nine months of age, the patient received HSCT from a 9/10 human leukocyte antigen (HLA)-matched unrelated donor.
HSCT resulted in a sustained reduction of lysosomal storage und bone metabolism markers. At six years of age, the patient showed normal cardiac function, partial respiratory insufficiency and moderate hepatomegaly, whereas skeletal manifestations had progressed. However, the patient could walk and maintained an overall good quality of life. Neurocognitive testing revealed a developmental quotient of 36%. The patient died at 6.6 years of age following a human metapneumovirus (hMPV) pneumonia.
The exact benefit remains unclear as current literature vastly lacks comparable data on MLII natural history patients. In order to evaluate experimental therapies, in-depth prospective studies and registries of untreated MLII patients are indispensable.
II型粘脂贮积症(MLII)是一种极为罕见的溶酶体贮积病,由溶酶体酶运输缺陷引起。临床特征为颅面畸形、心肺功能障碍、肝脾肿大、骨骼畸形和神经认知发育迟缓。通常在生命的第一个十年内死亡,且无治愈方法。少数MLII患者接受了造血干细胞移植(HSCT),但全面的随访数据极为匮乏。
一名3个月大的女性患者经确诊患有MLII,其基因存在c.2213C>A和c.2220_2221dup突变。9个月大时,该患者接受了来自9/10人类白细胞抗原(HLA)匹配的无关供体的HSCT。
HSCT导致溶酶体贮积和骨代谢标志物持续减少。6岁时,患者心脏功能正常,有部分呼吸功能不全和中度肝肿大,而骨骼表现有所进展。然而,患者能够行走,总体生活质量良好。神经认知测试显示发育商为36%。该患者在6.6岁时因人偏肺病毒(hMPV)肺炎死亡。
由于目前文献中极度缺乏关于MLII自然病史患者的可比数据,确切的益处尚不清楚。为了评估实验性疗法,对未经治疗的MLII患者进行深入的前瞻性研究和登记是必不可少的。