Department of Nephrology, Faculty of Medicine, Juntendo University, Tokyo, Japan.
Nephrology Unit, Internal Medicine, Saiyu Soka Hospital, 1-7-22 Matsubara, Soka, Saitama, 340-0041, Japan.
Clin J Gastroenterol. 2021 Aug;14(4):1175-1179. doi: 10.1007/s12328-021-01338-1. Epub 2021 Feb 5.
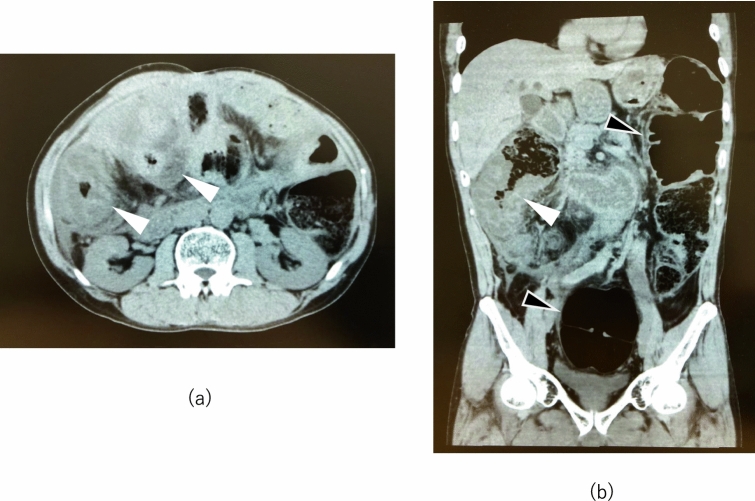
Hereditary angioedema due to C1-inhibitor deficiency (HAE-C1-INH) is a rare disease, which induces an acute attack of angioedema mediated by bradykinin. HAE-C1-INH can cause serious abdominal pain when severe edema develops in the gastrointestinal tract. However, because it takes a long time, 13.8 years on average in Japan, from the occurrence of the initial symptom to the diagnosis due to low awareness of the disease, undiagnosed HAE-C1-INH patients sometimes undergo unnecessary surgical procedures for severe abdominal pain. We herein present a 56-year-old patient with HAE-C1-INH, who underwent numerous abdominal operations. He frequently needed hospitalization with the administration of opioid due to severe abdominal pain. However, after he was accurately diagnosed with HAE-C1-INH at 55 years of age, he could start self-administration for an acute attack with icatibant, a selective bradykinin B2 receptor antagonist. Consequently, he did not need hospitalizing for ten months after the beginning of the treatment. A series of an accurate diagnosis and appropriate treatment for HAE-C1-INH improved his quality of life. Thus, HAE-C1-INH should be considered, when we meet patients with unidentified recurrent abdominal pain. This case highlights significance of an early diagnosis and appropriate treatment for HAE-C1-INH.
遗传性血管性水肿伴 C1 酯酶抑制剂缺乏症(HAE-C1-INH)是一种罕见疾病,由缓激肽介导导致急性血管性水肿发作。当胃肠道严重水肿时,HAE-C1-INH 可引起严重腹痛。然而,由于对该病的认识不足,从最初症状发生到确诊平均需要 13.8 年的时间,因此未确诊的 HAE-C1-INH 患者有时会因严重腹痛而接受不必要的手术。本文报告了一位 56 岁 HAE-C1-INH 患者,他接受了多次腹部手术。由于严重腹痛,他经常需要住院并使用阿片类药物。然而,在他 55 岁时被准确诊断为 HAE-C1-INH 后,他可以开始使用依替巴肽(一种选择性缓激肽 B2 受体拮抗剂)进行急性发作的自我治疗。因此,在开始治疗后的十个月内,他无需住院。一系列准确的诊断和适当的治疗改善了他的生活质量。因此,当我们遇到不明原因的复发性腹痛患者时,应考虑 HAE-C1-INH。本病例强调了早期诊断和适当治疗 HAE-C1-INH 的重要性。