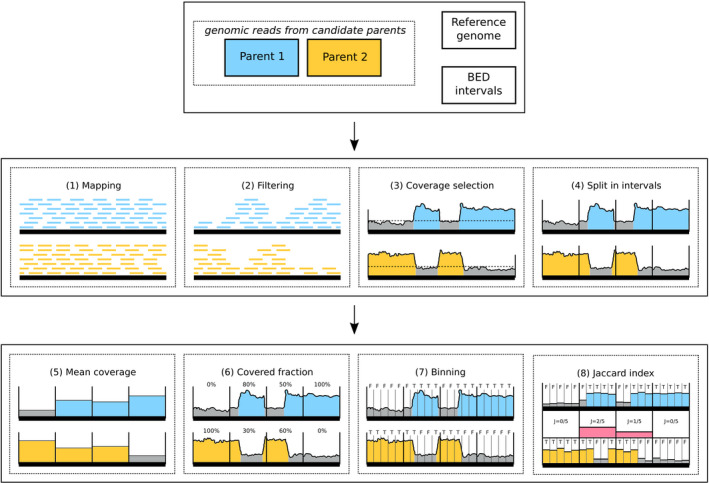
Department of Biotechnology, University of Natural Resources and Life Sciences (BOKU), Institute of Computational Biology, Muthgasse 18, Vienna, 1190, Austria.
Plant J. 2021 May;106(3):672-688. doi: 10.1111/tpj.15190. Epub 2021 Mar 24.
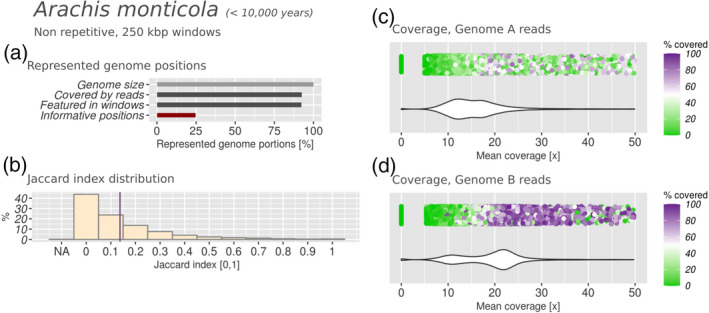
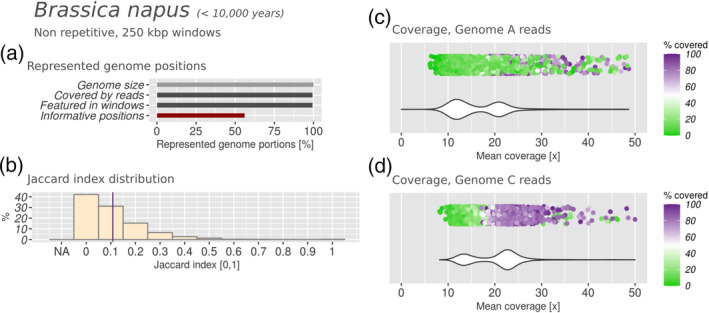
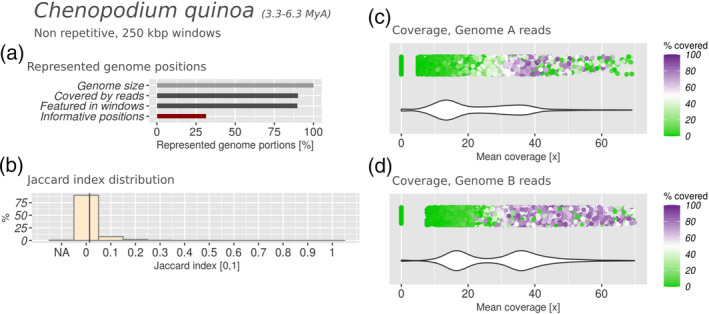
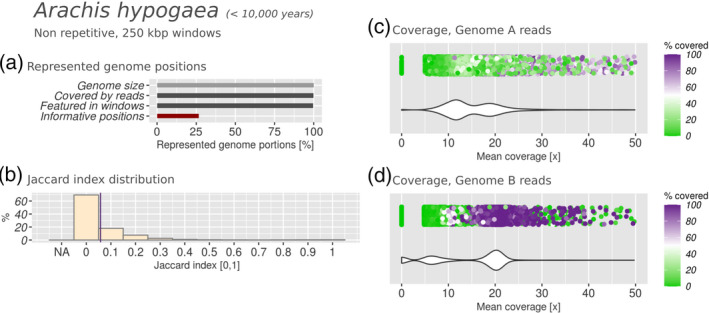
Polyploidization is a well-known speciation and adaptation mechanism. Traces of former polyploidization events were discovered within many genomes, and especially in plants. Allopolyploidization by interspecific hybridization between two species is common. Among hybrid plants, many are domesticated species of agricultural interest and many of their genomes and of their presumptive parents have been sequenced. Hybrid genomes remain challenging to analyse because of the presence of multiple subgenomes. The genomes of hybrids often undergo rearrangement and degradation over time. Based on 10 hybrid plant genomes from six different genera, with hybridization dating from 10,000 to 5 million years ago, we assessed subgenome degradation, subgenomic intermixing and biased subgenome fractionation. The restructuring of hybrid genomes does not proceed proportionally with the age of the hybrid. The oldest hybrids in our data set display completely different fates: whereas the subgenomes of the tobacco plant Nicotiana benthamiana are in an advanced stage of degradation, the subgenomes of quinoa (Chenopodium quinoa) are exceptionally well conserved by structure and sequence. We observed statistically significant biased subgenome fractionation in seven out of 10 hybrids, which had different ages and subgenomic intermixing levels. Hence, we conclude that no correlation exists between biased fractionation and subgenome intermixing. Lastly, domestication may encourage or hinder subgenome intermixing, depending on the evolutionary context. In summary, comparative analysis of hybrid genomes and their presumptive parents allowed us to determine commonalities and differences between their evolutionary fates. In order to facilitate the future analysis of further hybrid genomes, we automated the analysis steps within manticore, which is publicly available at https://github.com/MatteoSchiavinato/manticore.git.
多倍化是一种众所周知的物种形成和适应机制。在许多基因组中,特别是在植物中,发现了以前多倍化事件的痕迹。两个物种之间的种间杂交导致异源多倍体化很常见。在杂交植物中,许多是具有农业价值的驯化物种,它们的许多基因组及其假定亲本的基因组已经被测序。由于存在多个亚基因组,杂交基因组的分析仍然具有挑战性。随着时间的推移,杂交基因组经常发生重排和降解。基于来自六个不同属的 10 个杂交植物基因组,杂交时间从 10000 到 500 万年前不等,我们评估了亚基因组降解、亚基因组混合和偏亚基因组分离。杂交基因组的重组并不与杂交的年龄成比例进行。我们数据集中最古老的杂交种显示出完全不同的命运:虽然烟草 Nicotiana benthamiana 的亚基因组处于降解的高级阶段,但藜麦 (Chenopodium quinoa) 的亚基因组在结构和序列上都得到了异常良好的保护。我们在 10 个杂交种中的 7 个中观察到了具有统计学意义的偏亚基因组分离,这些杂交种的年龄和亚基因组混合水平不同。因此,我们得出的结论是,偏分离与亚基因组混合之间没有相关性。最后,根据进化背景的不同,驯化可能会促进或阻碍亚基因组的混合。总之,对杂交基因组及其假定亲本的比较分析确定了它们进化命运之间的共性和差异。为了方便未来对更多杂交基因组的分析,我们在 manticore 中自动化了分析步骤,manticore 可在 https://github.com/MatteoSchiavinato/manticore.git 上公开获取。