Ogata Kosuke, Chang Chih-Hsiang, Ishihama Yasushi
Department of Molecular and Cellular BioAnalysis, Graduate School of Pharmaceutical Sciences, Kyoto University, Sakyo-ku, Kyoto 606-8501, Japan.
Mass Spectrom (Tokyo). 2021;10:A0093. doi: 10.5702/massspectrometry.A0093. Epub 2021 Jan 30.
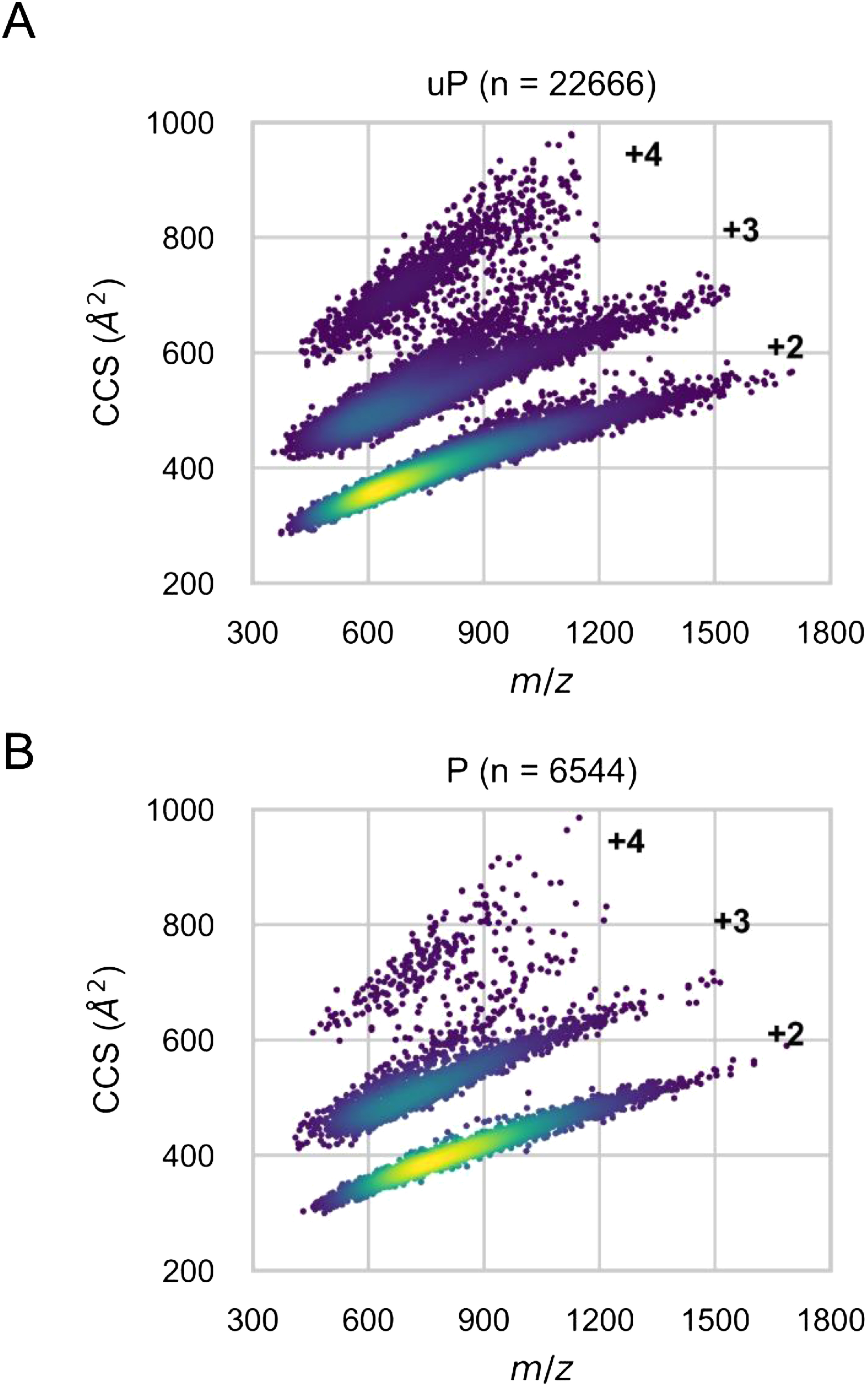
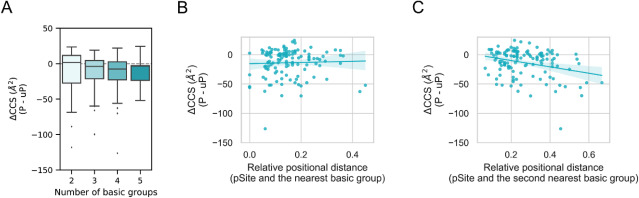
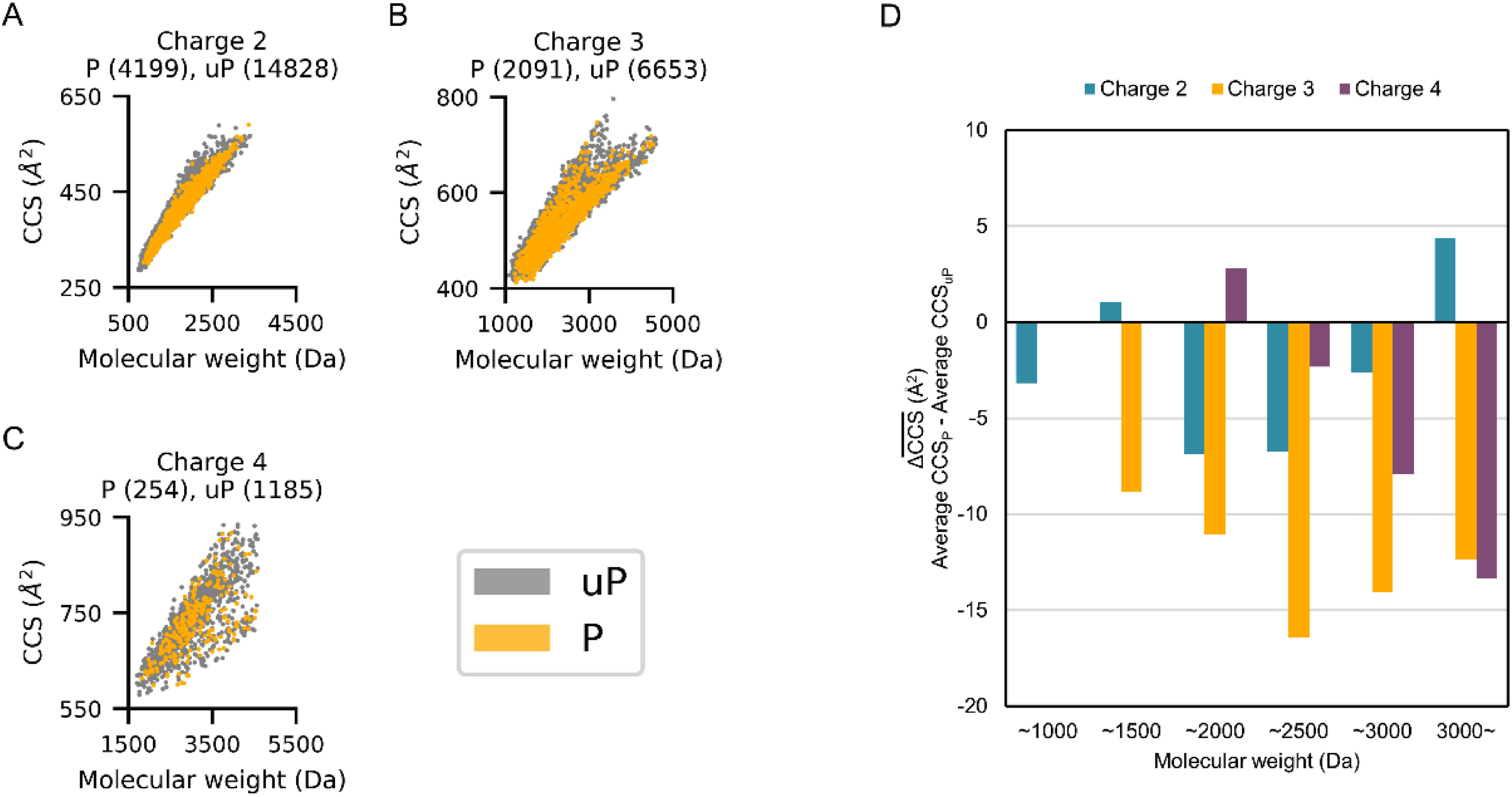
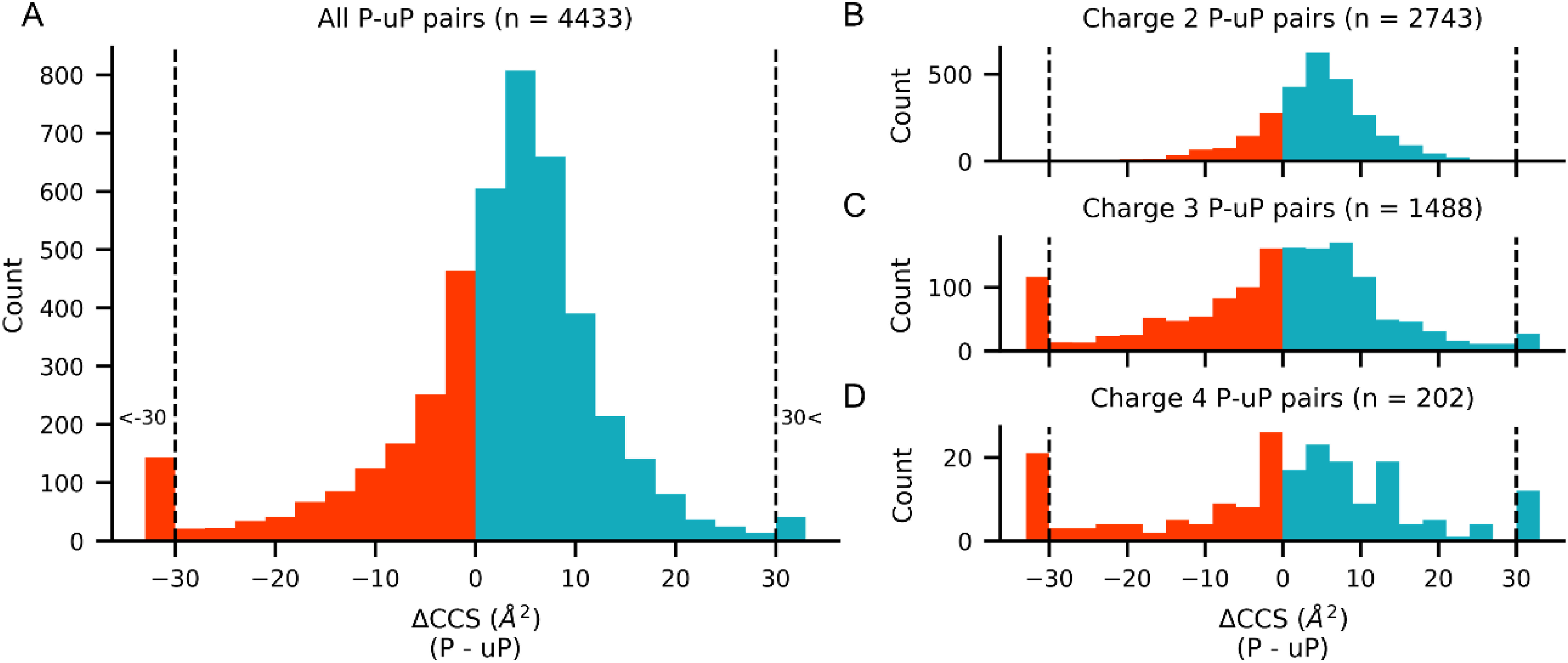
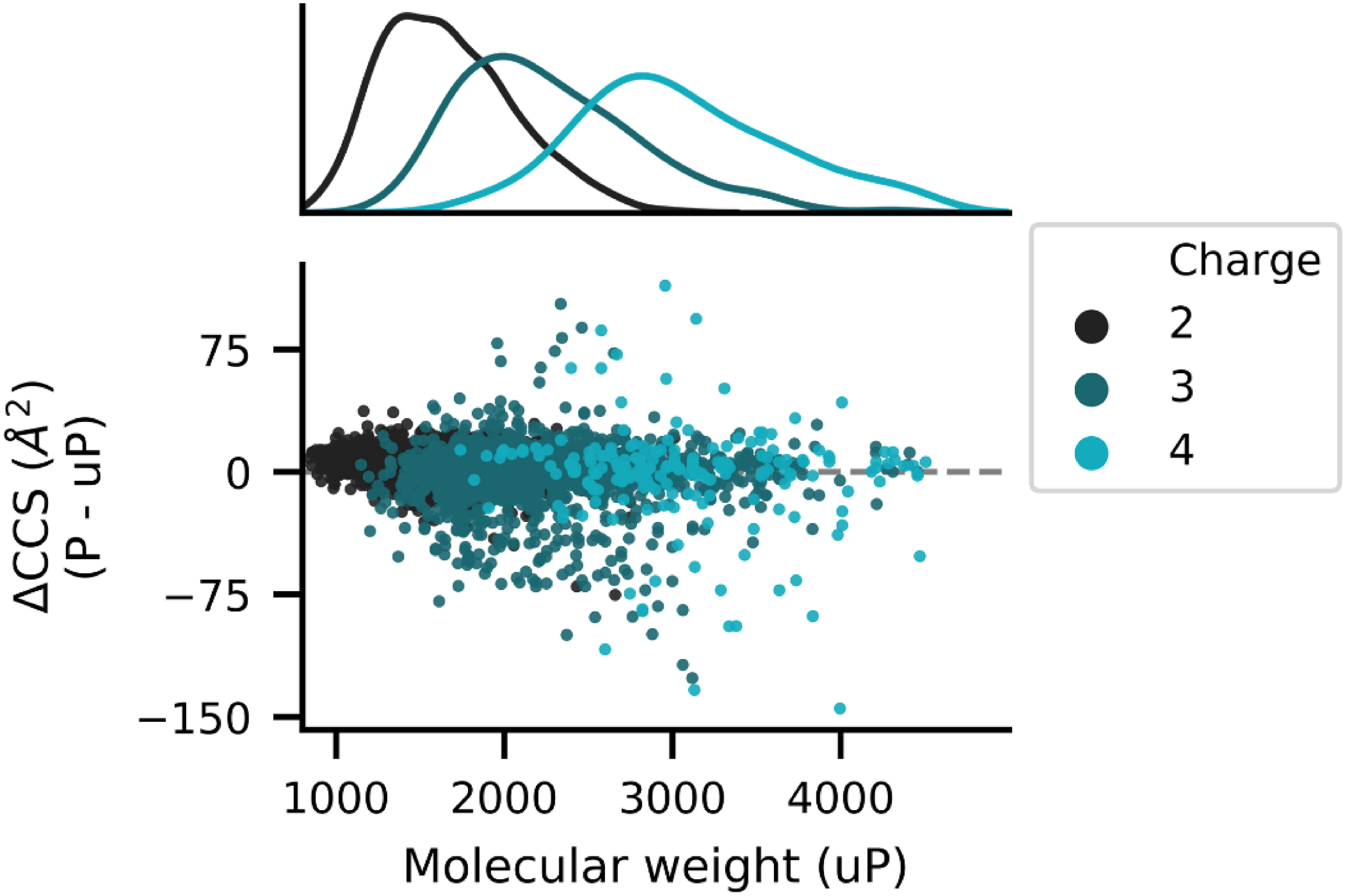
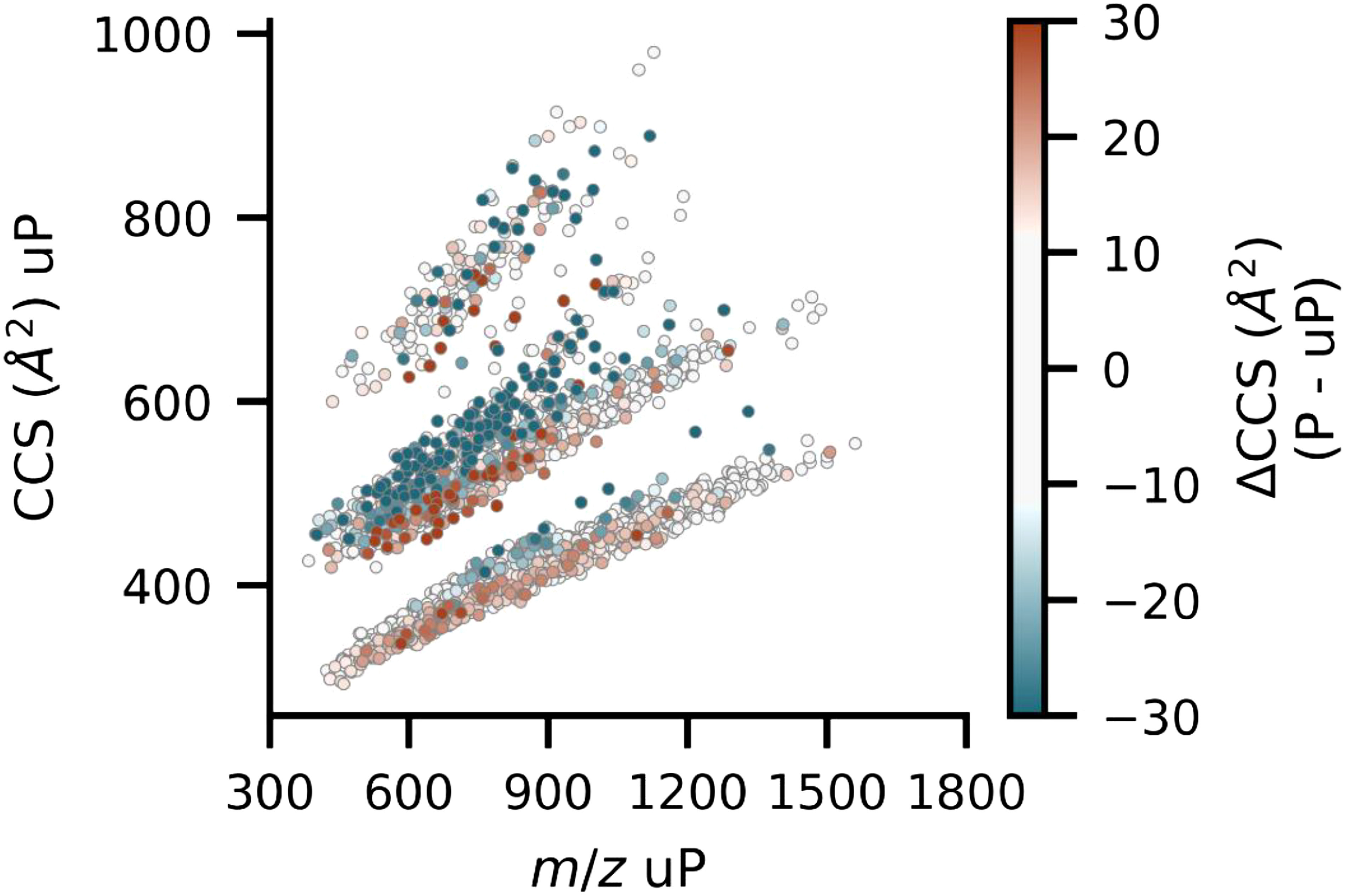
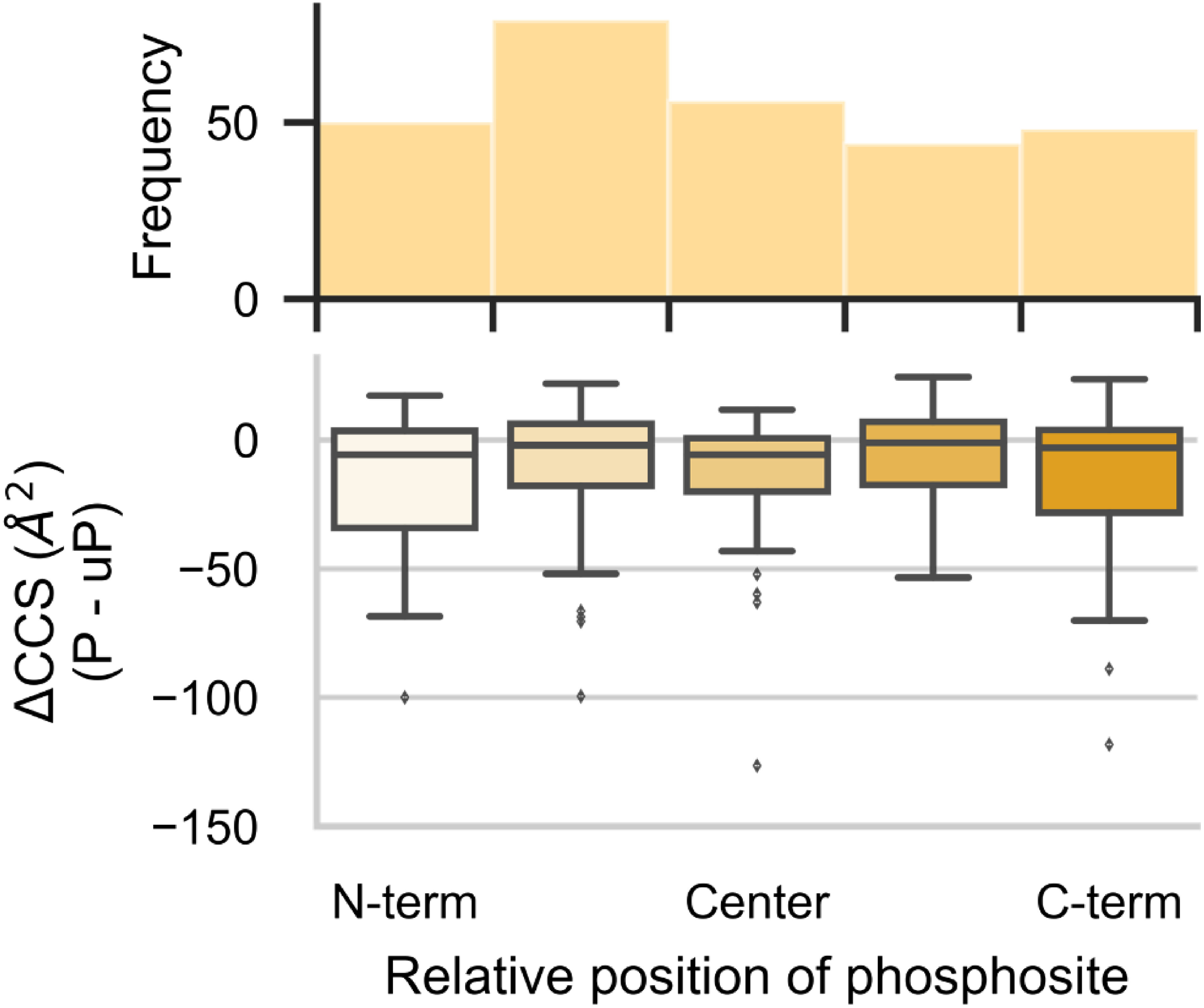
The insertion of ion mobility spectrometry (IMS) between LC and MS can improve peptide identification in both proteomics and phosphoproteomics by providing structural information that is complementary to LC and MS, because IMS separates ions on the basis of differences in their shapes and charge states. However, it is necessary to know how phosphate groups affect the peptide collision cross sections (CCS) in order to accurately predict phosphopeptide CCS values and to maximize the usefulness of IMS. In this work, we systematically characterized the CCS values of 4,433 pairs of mono-phosphopeptide and corresponding unphosphorylated peptide ions using trapped ion mobility spectrometry (TIMS). Nearly one-third of the mono-phosphopeptide ions evaluated here showed smaller CCS values than their unphosphorylated counterparts, even though phosphorylation results in a mass increase of 80 Da. Significant changes of CCS upon phosphorylation occurred mainly in structurally extended peptides with large numbers of basic groups, possibly reflecting intramolecular interactions between phosphate and basic groups.
在液相色谱(LC)和质谱(MS)之间插入离子淌度谱(IMS),可以通过提供与LC和MS互补的结构信息,改善蛋白质组学和磷酸化蛋白质组学中的肽段鉴定,因为IMS基于离子的形状和电荷状态差异来分离离子。然而,为了准确预测磷酸化肽段的碰撞截面(CCS)值并最大化IMS的效用,有必要了解磷酸基团如何影响肽段的碰撞截面。在这项工作中,我们使用捕集离子淌度谱(TIMS)系统地表征了4433对单磷酸化肽段和相应的未磷酸化肽段离子的CCS值。尽管磷酸化导致质量增加80 Da,但这里评估的近三分之一的单磷酸化肽段离子的CCS值比其未磷酸化的对应物小。磷酸化后CCS的显著变化主要发生在具有大量碱性基团的结构延伸肽段中,这可能反映了磷酸盐与碱性基团之间的分子内相互作用。