Department of Proteomics and Signal Transduction, Max Planck Institute of Biochemistry, Martinsried, Germany.
Department of Proteomics and Signal Transduction, Max Planck Institute of Biochemistry, Martinsried, Germany; Department of Functional Proteomics, Jena University Hospital, Jena, Germany.
Mol Cell Proteomics. 2022 Sep;21(9):100279. doi: 10.1016/j.mcpro.2022.100279. Epub 2022 Aug 6.
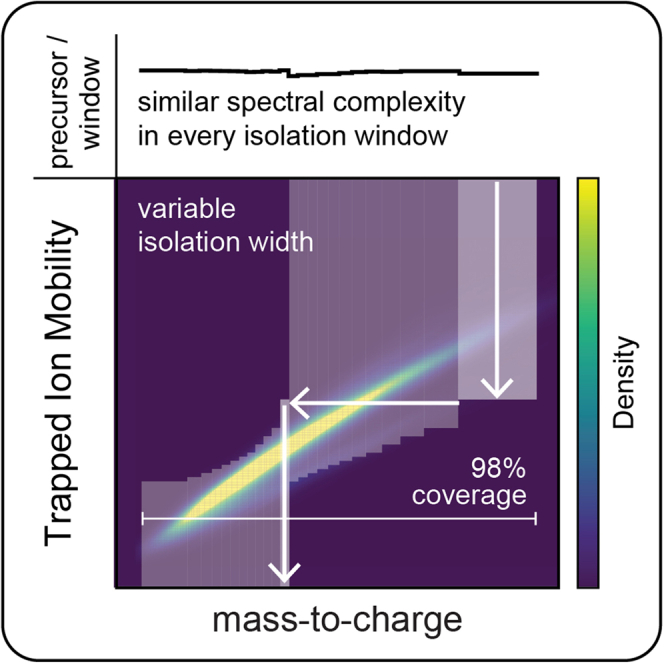
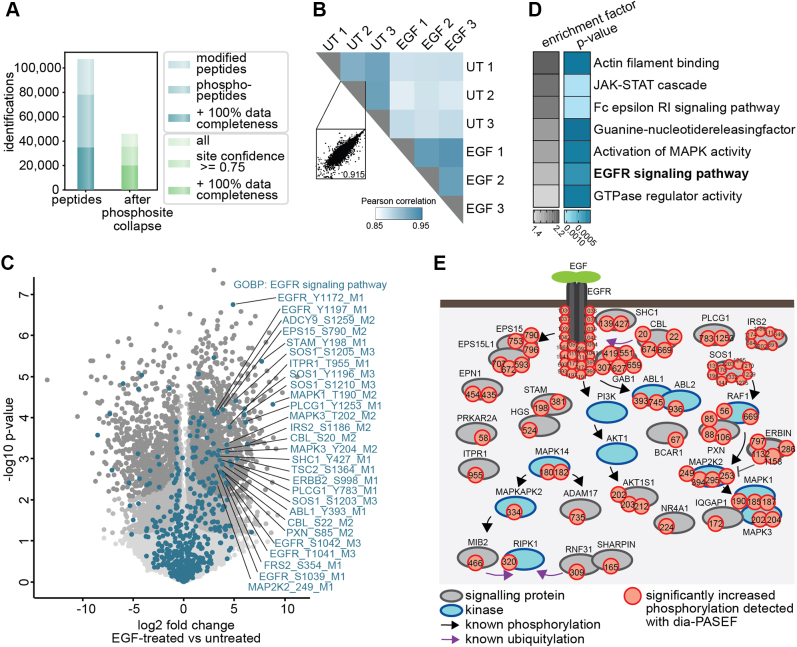
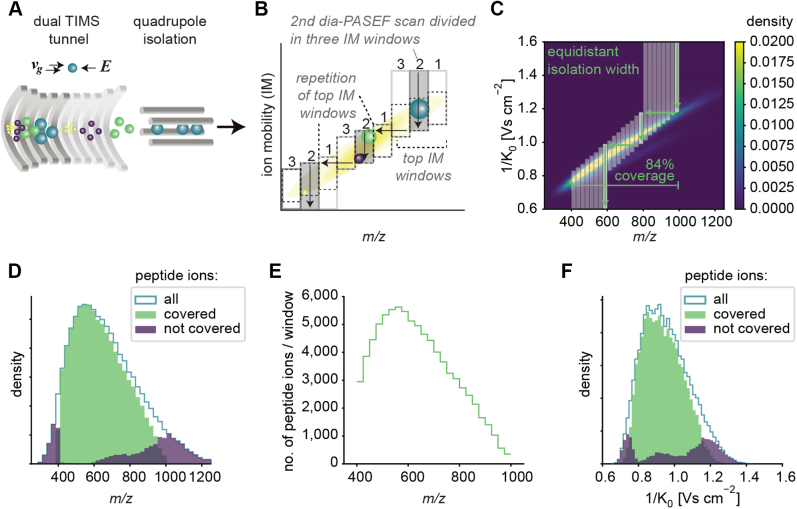
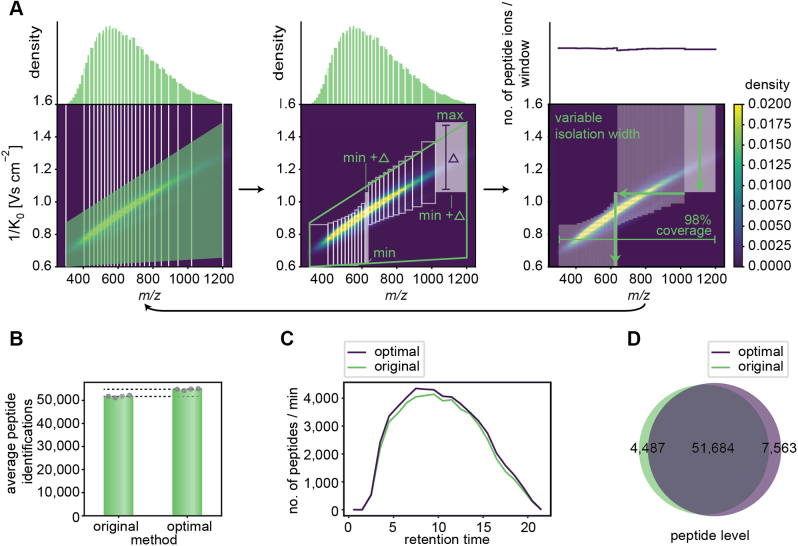
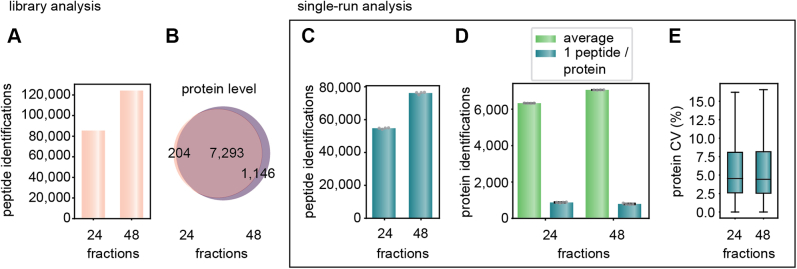
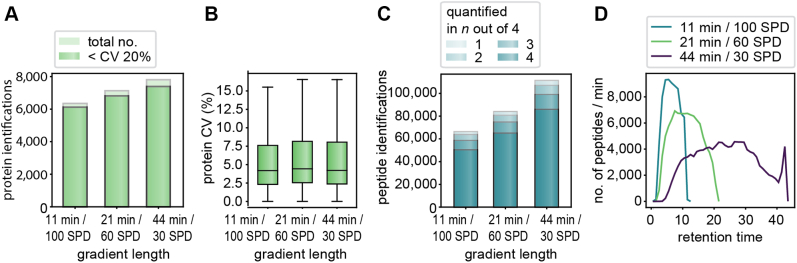
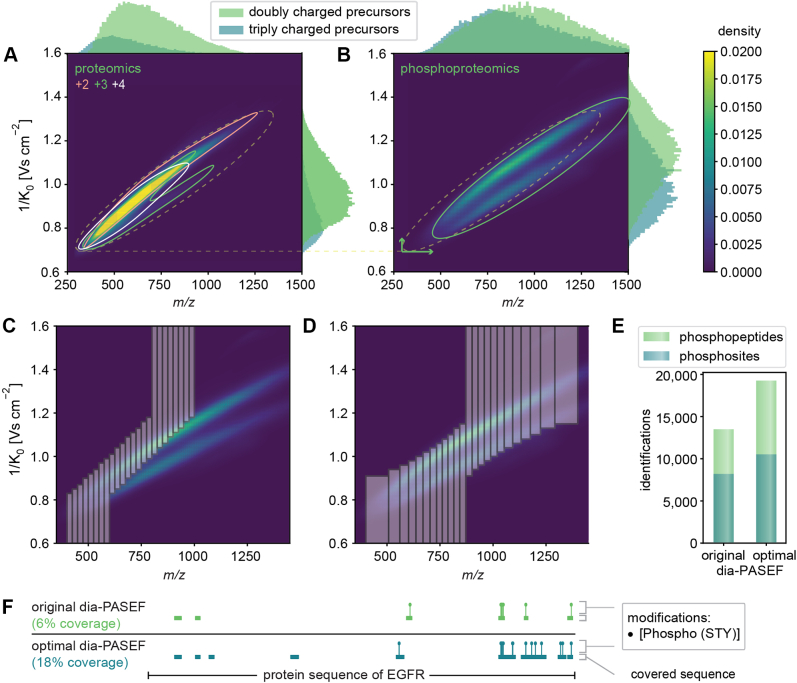
Data-independent acquisition (DIA) methods have become increasingly attractive in mass spectrometry-based proteomics because they enable high data completeness and a wide dynamic range. Recently, we combined DIA with parallel accumulation-serial fragmentation (dia-PASEF) on a Bruker trapped ion mobility (IM) separated quadrupole time-of-flight mass spectrometer. This requires alignment of the IM separation with the downstream mass selective quadrupole, leading to a more complex scheme for dia-PASEF window placement compared with DIA. To achieve high data completeness and deep proteome coverage, here we employ variable isolation windows that are placed optimally depending on precursor density in the m/z and IM plane. This is implemented in the freely available py_diAID (Python package for DIA with an automated isolation design) package. In combination with in-depth project-specific proteomics libraries and the Evosep liquid chromatography system, we reproducibly identified over 7700 proteins in a human cancer cell line in 44 min with quadruplicate single-shot injections at high sensitivity. Even at a throughput of 100 samples per day (11 min liquid chromatography gradients), we consistently quantified more than 6000 proteins in mammalian cell lysates by injecting four replicates. We found that optimal dia-PASEF window placement facilitates in-depth phosphoproteomics with very high sensitivity, quantifying more than 35,000 phosphosites in a human cancer cell line stimulated with an epidermal growth factor in triplicate 21 min runs. This covers a substantial part of the regulated phosphoproteome with high sensitivity, opening up for extensive systems-biological studies.
数据非依赖性采集(DIA)方法在基于质谱的蛋白质组学中变得越来越有吸引力,因为它们能够实现高数据完整性和宽动态范围。最近,我们在 Bruker 囚禁离子淌度(IM)分离四极杆飞行时间质谱仪上结合了 DIA 与平行累积-串行碎裂(dia-PASEF)。这需要将 IM 分离与下游质量选择四极杆对齐,这导致与 DIA 相比,dia-PASEF 窗口放置的方案更加复杂。为了实现高数据完整性和深度蛋白质组覆盖,我们在这里采用可变隔离窗口,这些窗口根据 m/z 和 IM 平面中的前体密度进行最佳放置。这是在免费提供的 py_diAID(用于 DIA 的 Python 包,具有自动化隔离设计)包中实现的。与深度项目特定蛋白质组学库和 Evosep 液相色谱系统相结合,我们在 44 分钟内以高灵敏度重复进样四次,在人癌细胞系中可重现地鉴定出超过 7700 种蛋白质。即使在 100 个样品/天(11 分钟的液相色谱梯度)的通量下,我们也通过注射四个重复,始终能够定量哺乳动物细胞裂解物中的 6000 多种蛋白质。我们发现,最佳的 dia-PASEF 窗口放置有助于进行非常灵敏的深度磷酸蛋白质组学,可在三重 21 分钟运行中对表皮生长因子刺激的人癌细胞系定量超过 35000 个磷酸化位点。这以高灵敏度涵盖了调控磷酸蛋白质组的大部分,为广泛的系统生物学研究开辟了道路。