Institute of Molecular Modeling and Simulation, University of Natural Resources and Life Sciences, Vienna A-1190, Austria.
Austrian Centre of Industrial Biotechnology, Petersgasse 14, Graz 8041, Austria.
J Chem Inf Model. 2021 Mar 22;61(3):1193-1203. doi: 10.1021/acs.jcim.0c01216. Epub 2021 Feb 11.
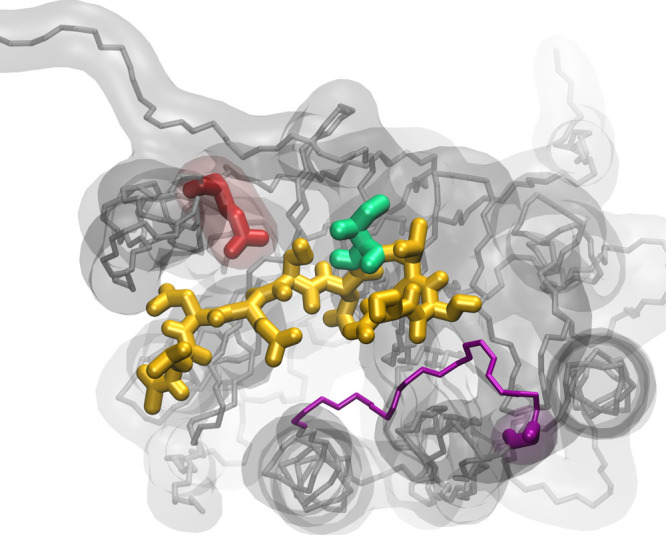
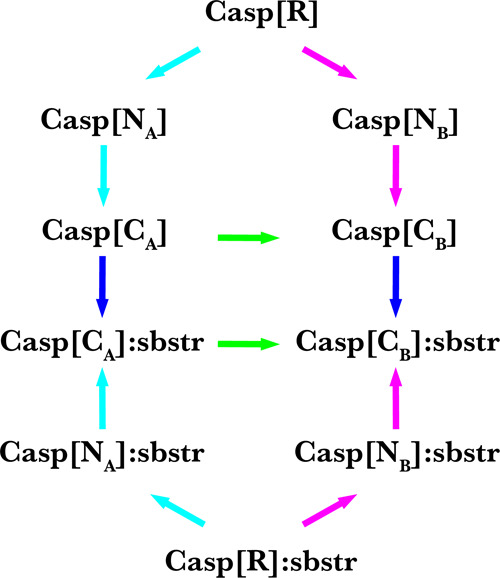
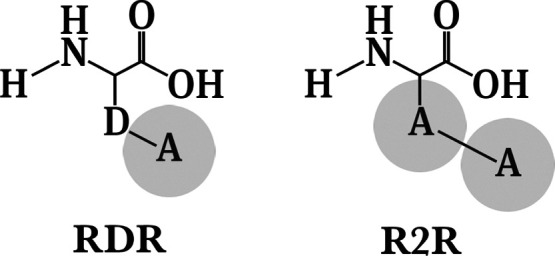
Rational-design methods have proven to be a valuable toolkit in the field of protein design. Numerical approaches such as free-energy calculations or QM/MM methods are fit to widen the understanding of a protein-sequence space but require large amounts of computational time and power. Here, we apply an efficient method for free-energy calculations that combines the one-step perturbation (OSP) with the third-power-fitting (TPF) approach. It is fit to calculate full free energies of binding from three different end states only. The nonpolar contribution to the free energies are calculated for a set of chosen amino acids from a single simulation of a judiciously chosen reference state. The electrostatic contributions, on the other hand, are predicted from simulations of the neutral and charged end states of the individual amino acids. We used this method to perform saturation mutagenesis of two sites in human Caspase-2. We calculated relative binding free energies toward two different substrates that differ in their P1' site and in their affinity toward the unmutated protease. Although being approximate, our approach showed very good agreement upon validation against experimental data. 76% of the predicted relative free energies of amino acid mutations was found to be true positives or true negatives. We observed that this method is fit to discriminate amino acid mutations because the rate of false negatives is very low (<1.5%). The approach works better for a substrate with medium/low affinity with a Matthews correlation coefficient (MCC) of 0.63, whereas for a substrate with very low affinity, the MCC was 0.38. In all cases, the combined TPF + OSP approach outperformed the linear interaction energy method.
理性设计方法已被证明是蛋白质设计领域的一种有价值的工具包。数值方法,如自由能计算或 QM/MM 方法,适合拓宽对蛋白质序列空间的理解,但需要大量的计算时间和能力。在这里,我们应用了一种有效的自由能计算方法,该方法将一步摄动(OSP)与三次拟合(TPF)方法相结合。它适合仅从三个不同的末端状态计算结合的全自由能。非极性贡献的自由能是从单个明智选择的参考状态的模拟中选择的一组氨基酸计算的。另一方面,静电贡献是从单个氨基酸的中性和带电末端状态的模拟中预测的。我们使用这种方法对人类 Caspase-2 的两个位点进行饱和突变。我们计算了两种不同底物的相对结合自由能,它们在 P1'位点和对未突变蛋白酶的亲和力方面有所不同。尽管是近似的,但我们的方法在与实验数据验证时显示出非常好的一致性。76%的预测氨基酸突变的相对自由能被证明是真阳性或真阴性。我们观察到这种方法适合区分氨基酸突变,因为假阴性的速度非常低(<1.5%)。对于亲和力适中/低的底物,该方法的效果更好,马修斯相关系数(MCC)为 0.63,而对于亲和力非常低的底物,MCC 为 0.38。在所有情况下,TPF+OSP 组合方法都优于线性相互作用能方法。