Department of Computer Science, West Bengal Education Service, Kolkata, India.
Department of Bioinformatics (IMG), University of Göttingen, 37077, Göttingen, Germany.
BMC Bioinformatics. 2021 Feb 11;22(1):64. doi: 10.1186/s12859-020-03918-3.
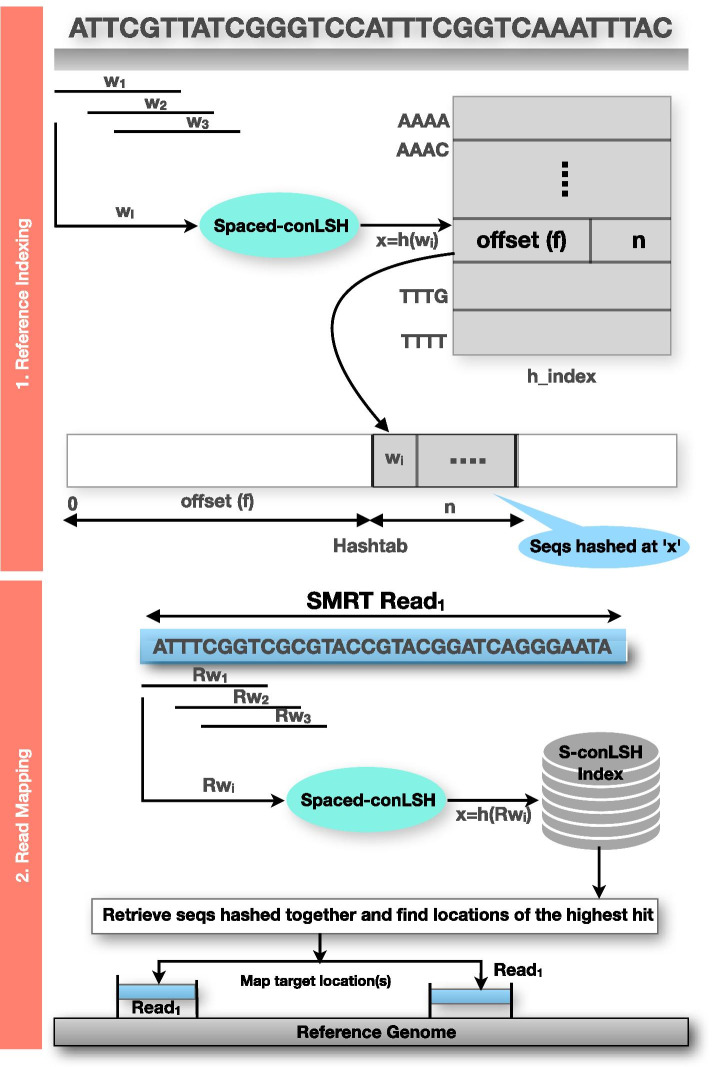
The advancement of SMRT technology has unfolded new opportunities of genome analysis with its longer read length and low GC bias. Alignment of the reads to their appropriate positions in the respective reference genome is the first but costliest step of any analysis pipeline based on SMRT sequencing. However, the state-of-the-art aligners often fail to identify distant homologies due to lack of conserved regions, caused by frequent genetic duplication and recombination. Therefore, we developed a novel alignment-free method of sequence mapping that is fast and accurate.
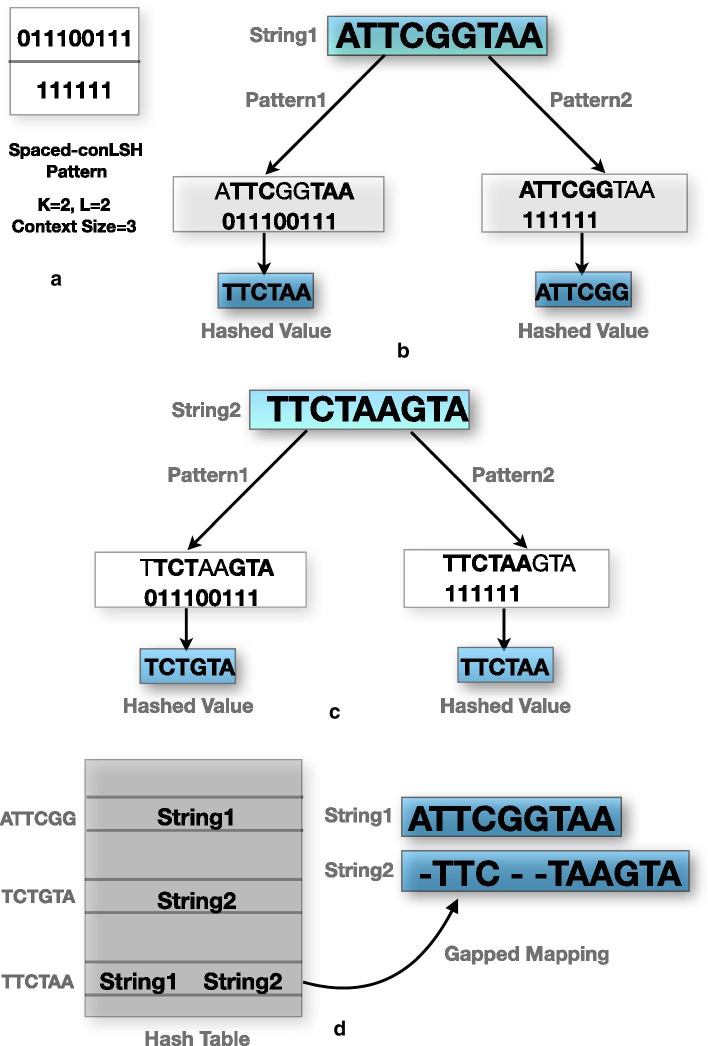
We present a new mapper called S-conLSH that uses Spaced context based Locality Sensitive Hashing. With multiple spaced patterns, S-conLSH facilitates a gapped mapping of noisy long reads to the corresponding target locations of a reference genome. We have examined the performance of the proposed method on 5 different real and simulated datasets. S-conLSH is at least 2 times faster than the recently developed method lordFAST. It achieves a sensitivity of 99%, without using any traditional base-to-base alignment, on human simulated sequence data. By default, S-conLSH provides an alignment-free mapping in PAF format. However, it has an option of generating aligned output as SAM-file, if it is required for any downstream processing.
S-conLSH is one of the first alignment-free reference genome mapping tools achieving a high level of sensitivity. The spaced-context is especially suitable for extracting distant similarities. The variable-length spaced-seeds or patterns add flexibility to the proposed algorithm by introducing gapped mapping of the noisy long reads. Therefore, S-conLSH may be considered as a prominent direction towards alignment-free sequence analysis.
SMRT 技术的发展为基因组分析带来了新的机遇,其具有更长的读取长度和低 GC 偏倚。在任何基于 SMRT 测序的分析管道中,将读取与相应参考基因组中的适当位置对齐是第一步,但也是最昂贵的步骤。然而,由于缺乏保守区域,最先进的对齐器经常无法识别远距离同源性,这是由于频繁的遗传重复和重组造成的。因此,我们开发了一种新的快速而准确的无比对序列映射方法。
我们提出了一种新的称为 S-conLSH 的映射器,它使用基于间隔上下文的局部敏感哈希。通过多个间隔模式,S-conLSH 可以实现嘈杂的长读取与参考基因组的相应目标位置的有间隙映射。我们已经在 5 个不同的真实和模拟数据集上检验了该方法的性能。S-conLSH 比最近开发的 lordFAST 方法至少快 2 倍。在人类模拟序列数据上,它在不使用任何传统的基于碱基的比对的情况下,实现了 99%的敏感性。默认情况下,S-conLSH 以 PAF 格式提供无比对映射。但是,如果需要进行任何下游处理,它具有生成对齐输出作为 SAM 文件的选项。
S-conLSH 是第一个实现高灵敏度的无比对参考基因组映射工具之一。间隔上下文特别适合提取远距离相似性。可变长度的间隔种子或模式通过引入嘈杂的长读取的有间隙映射,为所提出的算法增加了灵活性。因此,S-conLSH 可以被视为无比对序列分析的一个重要方向。