Muthiah Annapoorani, Housley Gary D, Klugmann Matthias, Fröhlich Dominik
Translational Neuroscience Facility and Department of Physiology, School of Medical Sciences, UNSW Sydney, Kensington, NSW, Australia.
Front Cell Neurosci. 2021 Jan 26;14:626610. doi: 10.3389/fncel.2020.626610. eCollection 2020.
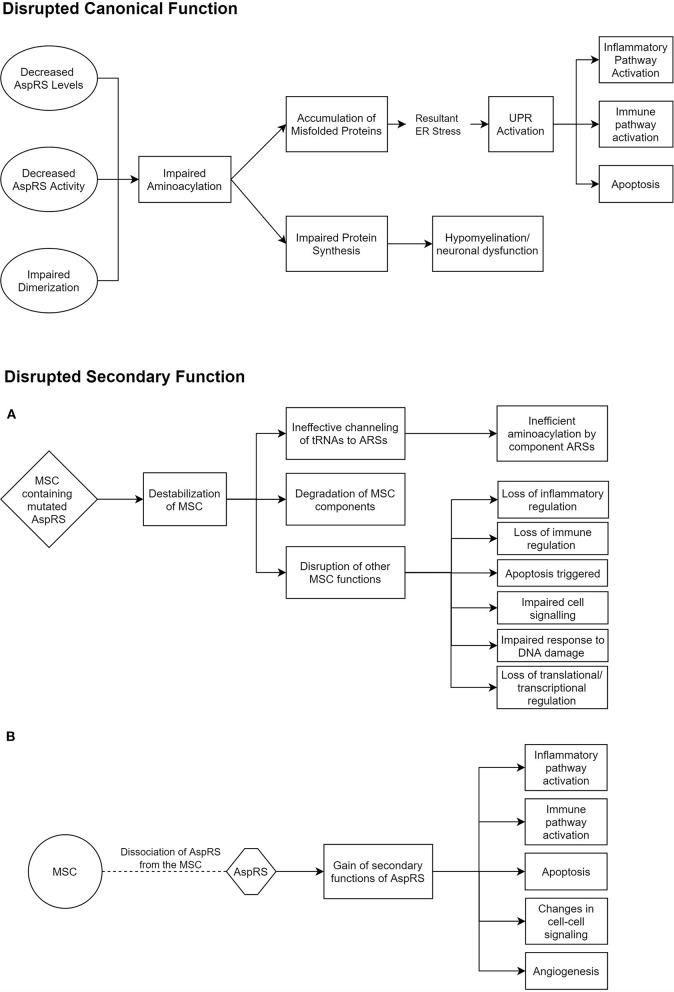
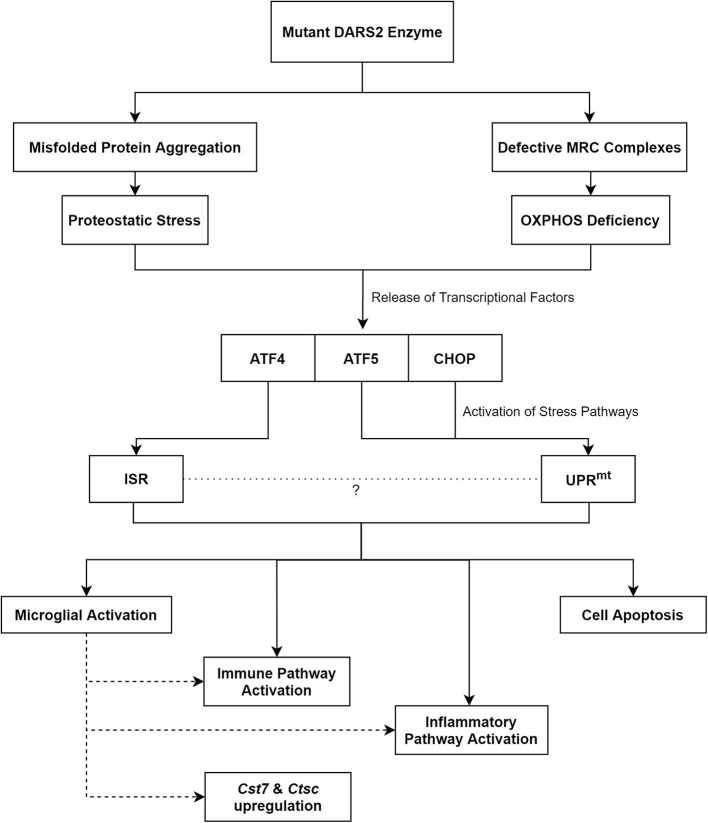
Aminoacyl-tRNA synthetases (ARSs) accurately charge tRNAs with their respective amino acids. As such, they are vital for the initiation of cytosolic and mitochondrial protein translation. These enzymes have become increasingly scrutinized in recent years for their role in neurodegenerative disorders caused by the mutations of ARS-encoding genes. This review focuses on two such genes- and -which encode cytosolic and mitochondrial aspartyl-tRNA synthetases, and the clinical conditions associated with mutations of these genes. We also describe attempts made at modeling these conditions in mice, which have both yielded important mechanistic insights. eukoencephalopathy with rainstem and pinal cord involvement and actate elevation (LBSL) is a disease caused by a range of mutations in the gene, initially identified in 2003. Ten years later, ypomyelination with rainstem and pinal cord involvement and eg spasticity (HBSL), caused by mutations of cytosolic , was discovered. Multiple parallels have been drawn between the two conditions. The Magnetic Resonance Imaging (MRI) patterns are strikingly similar, but still set these two conditions apart from other leukodystrophies. Clinically, both conditions are characterized by lower limb spasticity, often associated with other pyramidal signs. However, perhaps due to earlier detection, a wider range of symptoms, including peripheral neuropathy, as well as visual and hearing changes have been described in LBSL patients. Both HBSL and LBSL are spectrum disorders lacking genotype to phenotype correlation. While the fatal phenotype of or single gene deletion mouse mutants revealed that the two enzymes lack functional redundancy, further pursuit of disease modeling are required to shed light onto the underlying disease mechanism, and enable examination of experimental treatments, including gene therapies.
氨酰 - tRNA合成酶(ARSs)能将各自对应的氨基酸准确地加载到tRNA上。因此,它们对于胞质和线粒体蛋白质翻译的起始至关重要。近年来,这些酶因其在由ARS编码基因突变引起的神经退行性疾病中的作用而受到越来越多的审视。本综述聚焦于两个这样的基因——它们分别编码胞质和线粒体天冬氨酰 - tRNA合成酶,以及与这些基因突变相关的临床病症。我们还描述了在小鼠中对这些病症进行建模的尝试,这两者都产生了重要的机制性见解。伴脑干和脊髓受累及乳酸升高的脑白质病(LBSL)是一种由该基因一系列突变引起的疾病,于2003年首次被发现。十年后,发现了由胞质的突变导致的伴脑干和脊髓受累及痉挛的低髓鞘形成(HBSL)。这两种病症之间存在多个相似之处。磁共振成像(MRI)模式惊人地相似,但仍使这两种病症与其他脑白质营养不良区分开来。在临床上,这两种病症都以下肢痉挛为特征,通常伴有其他锥体束征。然而,也许是由于早期检测,LBSL患者中描述了更广泛的症状,包括周围神经病变以及视觉和听觉变化。HBSL和LBSL都是缺乏基因型与表型相关性的谱系障碍。虽然或单基因缺失小鼠突变体的致死表型表明这两种酶缺乏功能冗余,但需要进一步进行疾病建模以阐明潜在的疾病机制,并能够检验包括基因疗法在内的实验性治疗方法。