Marchet Camille, Morisse Pierre, Lecompte Lolita, Lefebvre Arnaud, Lecroq Thierry, Peterlongo Pierre, Limasset Antoine
Univ Rennes, CNRS, Inria, IRISA-UMR 6074, F-35000 Rennes, France.
Univ. Lille, CNRS, UMR 9189 - CRIStAL, 59655 Villeneuve-d'Ascq, France.
NAR Genom Bioinform. 2019 Nov 14;2(1):lqz015. doi: 10.1093/nargab/lqz015. eCollection 2020 Mar.
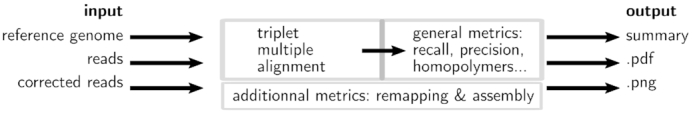
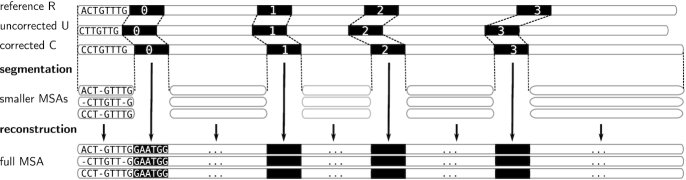
The error rates of third-generation sequencing data have been capped >5%, mainly containing insertions and deletions. Thereby, an increasing number of diverse long reads correction methods have been proposed. The quality of the correction has huge impacts on downstream processes. Therefore, developing methods allowing to evaluate error correction tools with precise and reliable statistics is a crucial need. These evaluation methods rely on costly alignments to evaluate the quality of the corrected reads. Thus, key features must allow the fast comparison of different tools, and scale to the increasing length of the long reads. Our tool, ELECTOR, evaluates long reads correction and is directly compatible with a wide range of error correction tools. As it is based on multiple sequence alignment, we introduce a new algorithmic strategy for alignment segmentation, which enables us to scale to large instances using reasonable resources. To our knowledge, we provide the unique method that allows producing reproducible correction benchmarks on the latest ultra-long reads (>100 k bases). It is also faster than the current state-of-the-art on other datasets and provides a wider set of metrics to assess the read quality improvement after correction. ELECTOR is available on GitHub (https://github.com/kamimrcht/ELECTOR) and Bioconda.
第三代测序数据的错误率一直高于5%,主要包含插入和缺失。因此,越来越多不同的长读段校正方法被提出。校正的质量对下游流程有巨大影响。所以,开发能够用精确且可靠的统计数据评估错误校正工具的方法是一项迫切需求。这些评估方法依赖于代价高昂的比对来评估校正后读段的质量。因此,关键特性必须允许对不同工具进行快速比较,并能适应长读段长度不断增加的情况。我们的工具ELECTOR可评估长读段校正,并且直接与多种错误校正工具兼容。由于它基于多序列比对,我们引入了一种新的比对分割算法策略,这使我们能够使用合理资源扩展到大型实例。据我们所知,我们提供了唯一一种能够在最新的超长读段(>100 k碱基)上生成可重复校正基准的方法。它在其他数据集上也比当前的最先进方法更快,并提供了更广泛的指标来评估校正后读段质量的提升。ELECTOR可在GitHub(https://github.com/kamimrcht/ELECTOR)和Bioconda上获取。