Edgar Robert
None, Corte Madera, CA, USA.
PeerJ. 2021 Feb 5;9:e10805. doi: 10.7717/peerj.10805. eCollection 2021.
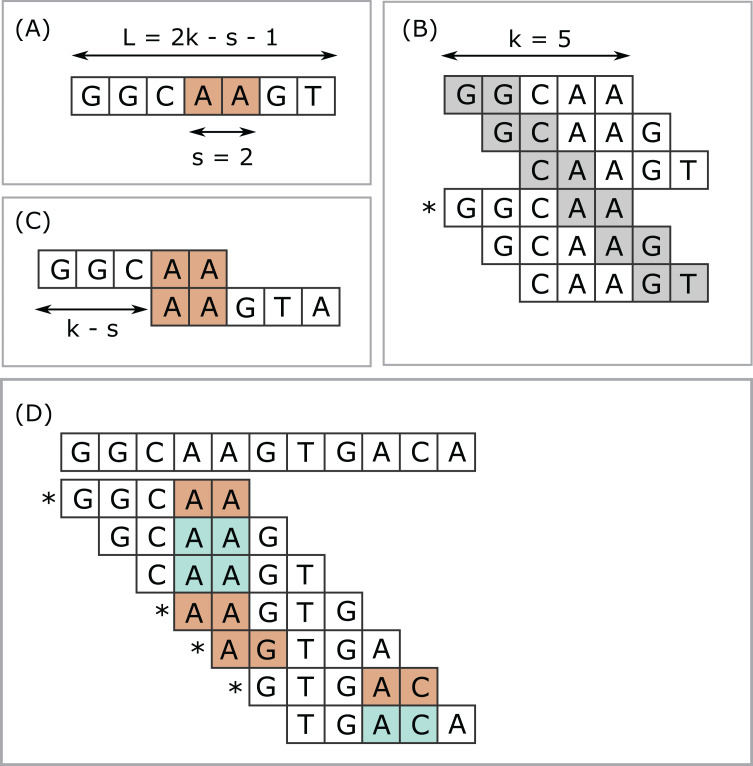
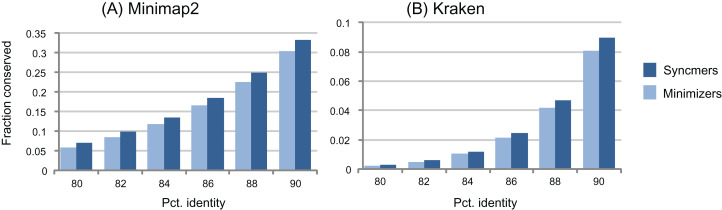
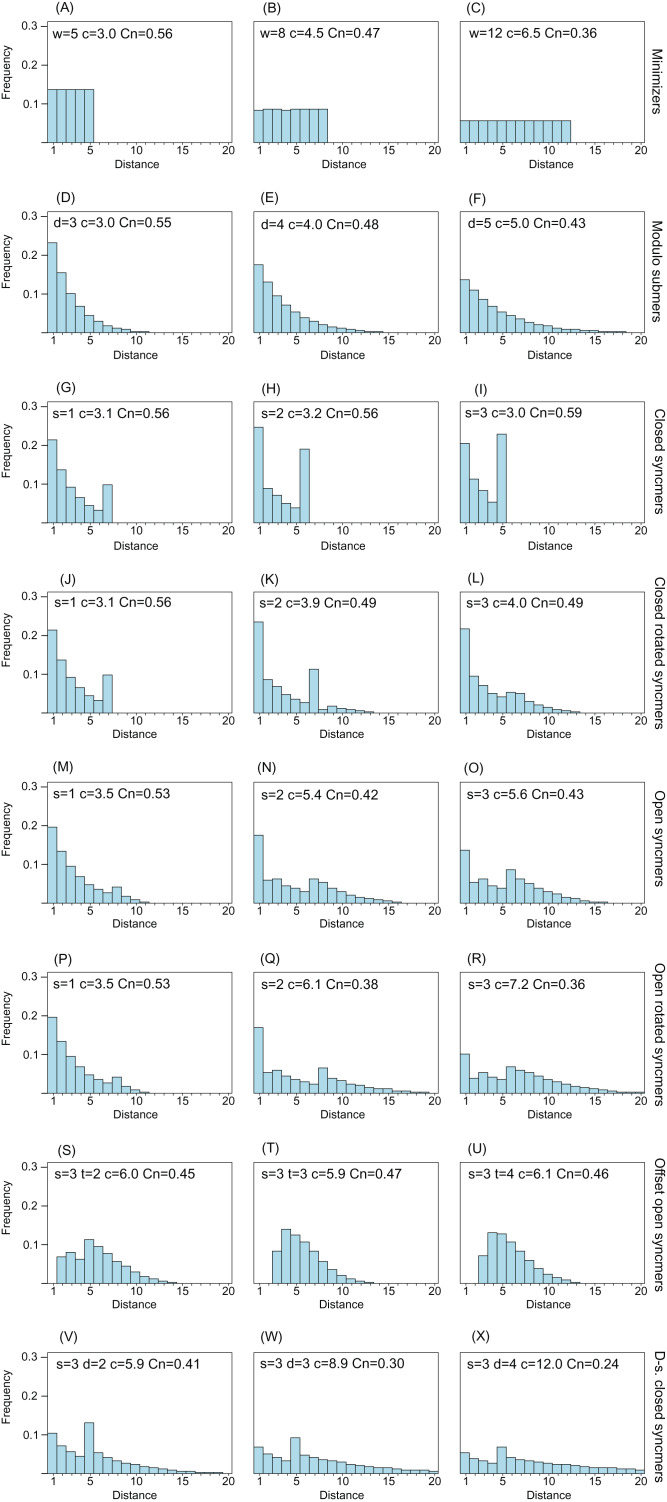
Minimizers are widely used to select subsets of fixed-length substrings (-mers) from biological sequences in applications ranging from read mapping to taxonomy prediction and indexing of large datasets. The minimizer of a string of consecutive -mers is the -mer with smallest value according to an ordering of all -mers. Syncmers are defined here as a family of alternative methods which select -mers by inspecting the position of the smallest-valued substring of length < within the -mer. For example, a closed syncmer is selected if its smallest -mer is at the start or end of the -mer. At least one closed syncmer must be found in every window of length ( - ) -mers. Unlike a minimizer, a syncmer is identified by its sequence alone, and is therefore synchronized in the following sense: if a given -mer is selected from one sequence, it will also be selected from any other sequence. Also, minimizers can be deleted by mutations in flanking sequence, which cannot happen with syncmers. Experiments on minimizers with parameters used in the minimap2 read mapper and Kraken taxonomy prediction algorithm respectively show that syncmers can simultaneously achieve both lower density and higher conservation compared to minimizers.
最小化器在从读取映射到分类预测以及大型数据集索引等各种应用中,被广泛用于从生物序列中选择固定长度子串(k-mers)的子集。一串连续k-mers的最小化器是根据所有k-mers的排序具有最小值的k-mer。同步k-mer在此被定义为一类替代方法,它通过检查长度为l < k的最小价值子串在k-mer内的位置来选择k-mers。例如,如果其最小k-mer在k-mer的开头或结尾,则选择一个封闭同步k-mer。在每个长度为(k - l)个k-mer的窗口中必须至少找到一个封闭同步k-mer。与最小化器不同,同步k-mer仅由其序列识别,因此在以下意义上是同步的:如果从一个序列中选择了给定的k-mer,那么它也将从任何其他序列中被选择。此外,最小化器可能会因侧翼序列中的突变而被删除,而同步k-mer不会出现这种情况。分别使用minimap2读取映射器和Kraken分类预测算法中使用的参数对最小化器进行的实验表明,与最小化器相比,同步k-mer可以同时实现更低的密度和更高的保守性。