Department Proteomics and Signal Transduction, Max Planck Institute of Biochemistry, Martinsried, Germany.
Functional Proteomics, Jena University Hospital, Jena, Germany.
Nat Commun. 2021 Feb 19;12(1):1185. doi: 10.1038/s41467-021-21352-8.
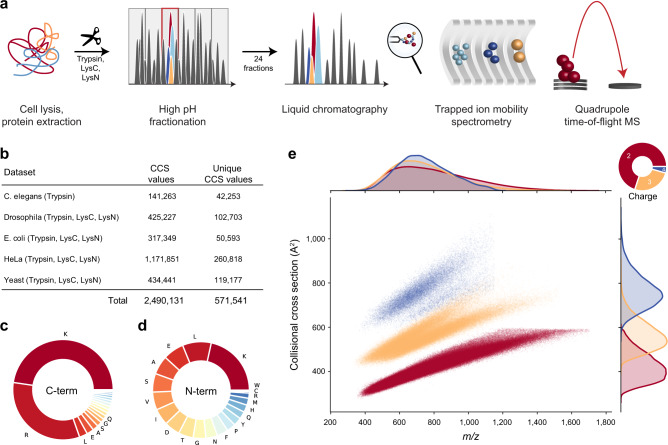
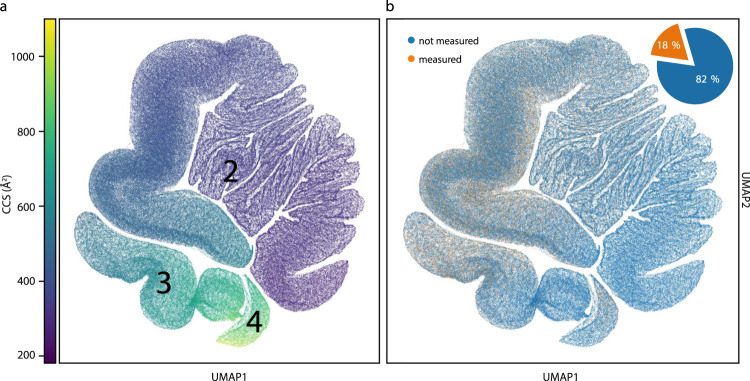
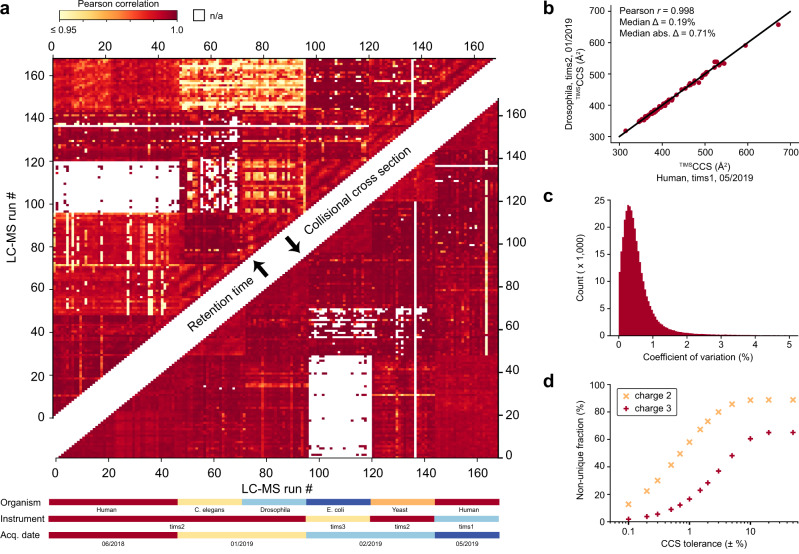
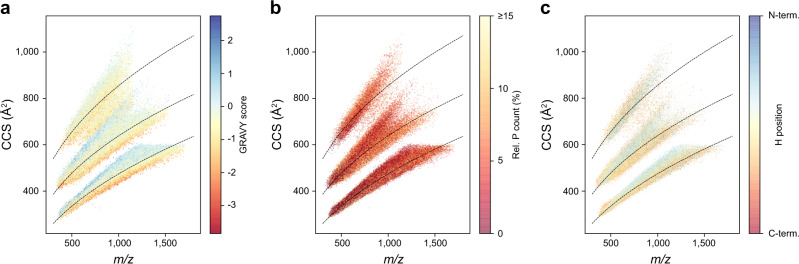
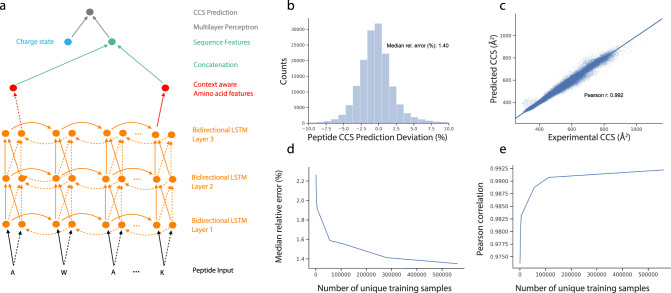
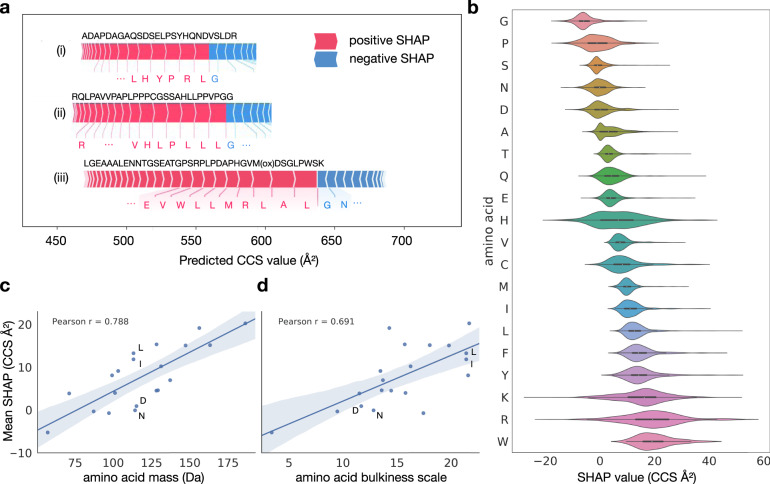
The size and shape of peptide ions in the gas phase are an under-explored dimension for mass spectrometry-based proteomics. To investigate the nature and utility of the peptide collisional cross section (CCS) space, we measure more than a million data points from whole-proteome digests of five organisms with trapped ion mobility spectrometry (TIMS) and parallel accumulation-serial fragmentation (PASEF). The scale and precision (CV < 1%) of our data is sufficient to train a deep recurrent neural network that accurately predicts CCS values solely based on the peptide sequence. Cross section predictions for the synthetic ProteomeTools peptides validate the model within a 1.4% median relative error (R > 0.99). Hydrophobicity, proportion of prolines and position of histidines are main determinants of the cross sections in addition to sequence-specific interactions. CCS values can now be predicted for any peptide and organism, forming a basis for advanced proteomics workflows that make full use of the additional information.
肽离子在气相中的大小和形状是基于质谱的蛋白质组学中一个探索不足的维度。为了研究肽碰撞截面(CCS)空间的性质和用途,我们使用离子阱迁移谱(TIMS)和并行累积-串联碎裂(PASEF)技术对五种生物体的全蛋白质酶解物进行了超过 100 万个数据点的测量。我们的数据的规模和精度(CV<1%)足以训练一个深度递归神经网络,该网络仅根据肽序列就能准确预测 CCS 值。对合成 ProteomeTools 肽的横截面预测验证了该模型的中位数相对误差在 1.4%以内(R>0.99)。除了序列特异性相互作用之外,疏水性、脯氨酸比例和组氨酸位置也是横截面的主要决定因素。现在可以预测任何肽和生物体的 CCS 值,为充分利用附加信息的高级蛋白质组学工作流程奠定了基础。