Wessex Clinical Genetics Service, Princess Anne Hospital, University Hospital Southampton NHS Foundation Trust, Coxford Rd, Southampton, SO165YA, UK.
European Xenopus Resource Centre, University of Portsmouth School of Biological Sciences, King Henry Building, King Henry I Street, Portsmouth, PO1 2DY, UK.
Genome Med. 2021 Feb 25;13(1):34. doi: 10.1186/s13073-021-00850-w.
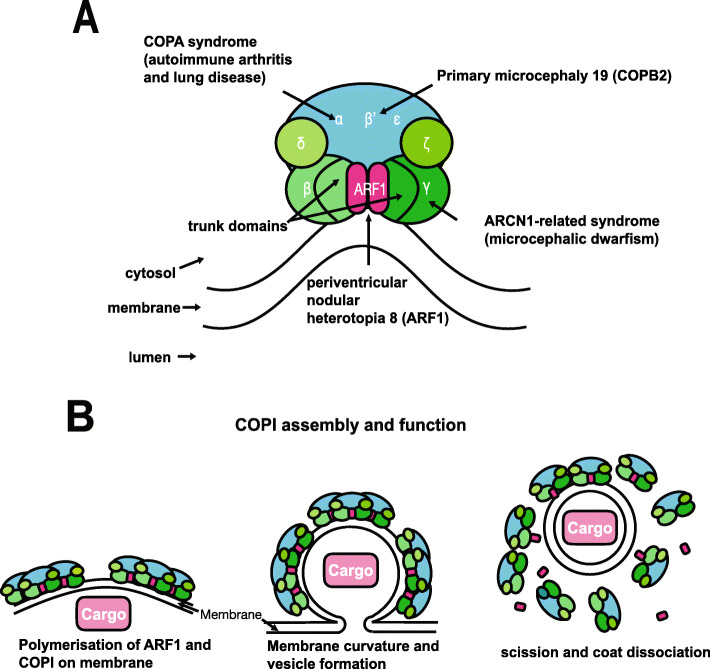
Coat protein complex 1 (COPI) is integral in the sorting and retrograde trafficking of proteins and lipids from the Golgi apparatus to the endoplasmic reticulum (ER). In recent years, coat proteins have been implicated in human diseases known collectively as "coatopathies".
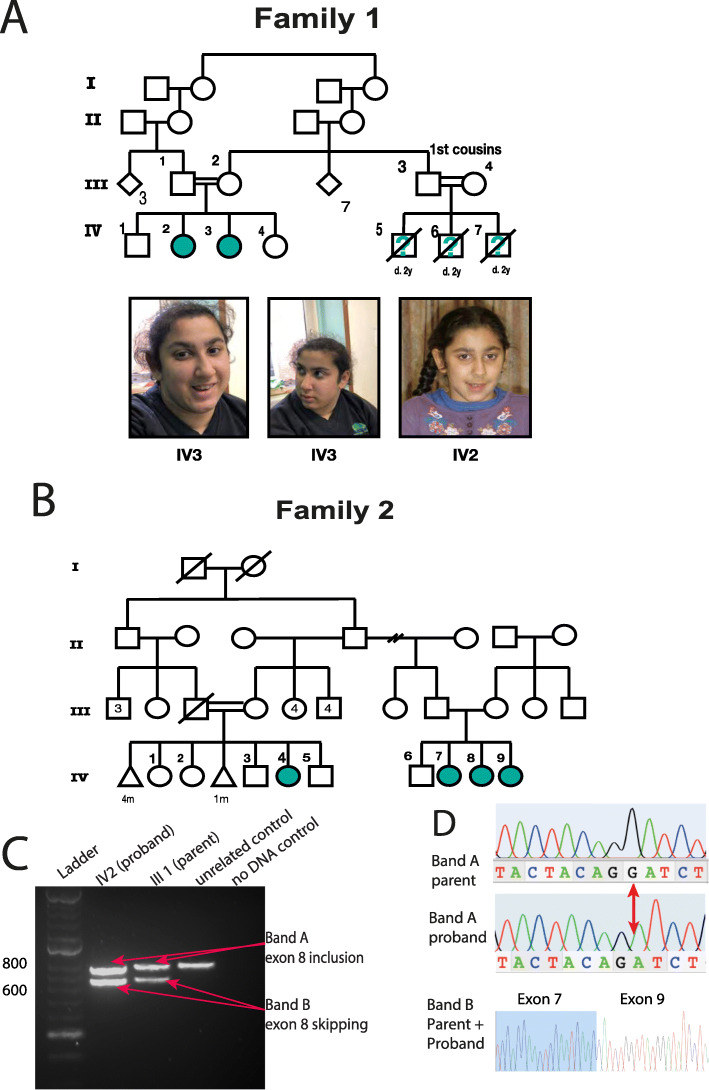
Whole exome or genome sequencing of two families with a neuro-developmental syndrome, variable microcephaly and cataracts revealed biallelic variants in COPB1, which encodes the beta-subunit of COPI (β-COP). To investigate Family 1's splice donor site variant, we undertook patient blood RNA studies and CRISPR/Cas9 modelling of this variant in a homologous region of the Xenopus tropicalis genome. To investigate Family 2's missense variant, we studied cellular phenotypes of human retinal epithelium and embryonic kidney cell lines transfected with a COPB1 expression vector into which we had introduced Family 2's mutation.
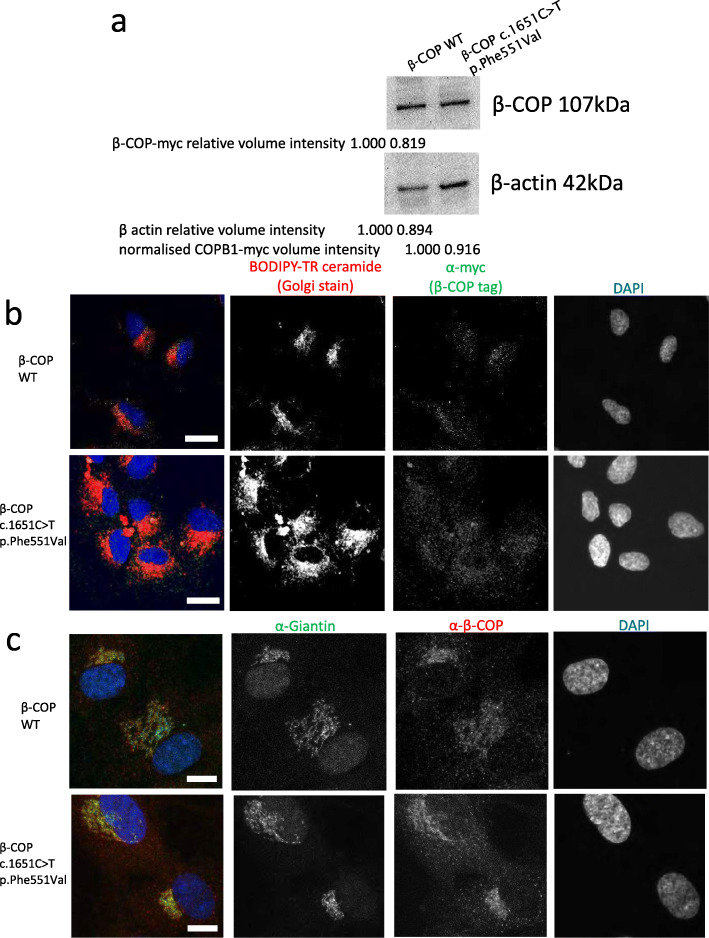
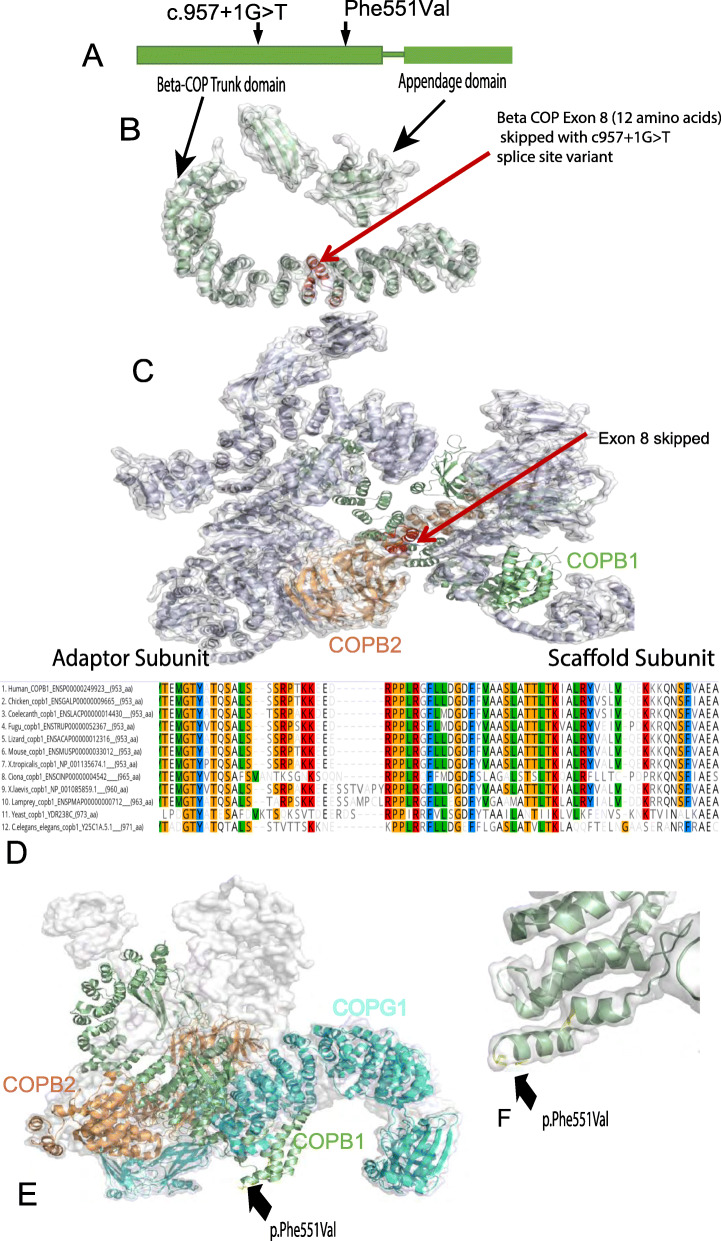
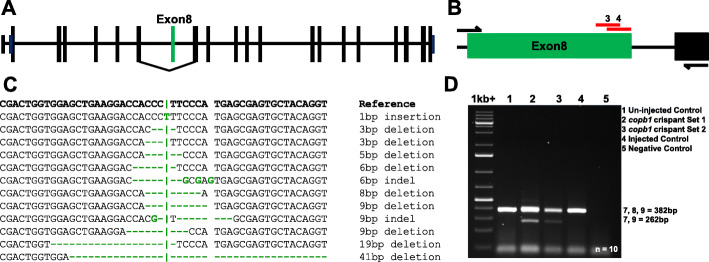
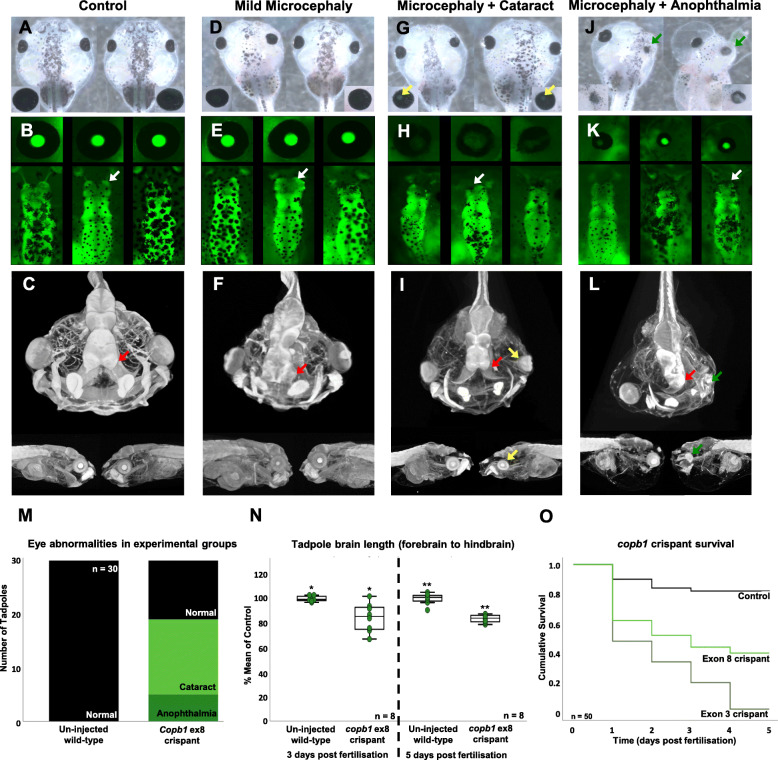
We present a new recessive coatopathy typified by severe developmental delay and cataracts and variable microcephaly. A homozygous splice donor site variant in Family 1 results in two aberrant transcripts, one of which causes skipping of exon 8 in COPB1 pre-mRNA, and a 36 amino acid in-frame deletion, resulting in the loss of a motif at a small interaction interface between β-COP and β'-COP. Xenopus tropicalis animals with a homologous mutation, introduced by CRISPR/Cas9 genome editing, recapitulate features of the human syndrome including microcephaly and cataracts. In vitro modelling of the COPB1 c.1651T>G p.Phe551Val variant in Family 2 identifies defective Golgi to ER recycling of this mutant β-COP, with the mutant protein being retarded in the Golgi.
This adds to the growing body of evidence that COPI subunits are essential in brain development and human health and underlines the utility of exome and genome sequencing coupled with Xenopus tropicalis CRISPR/Cas modelling for the identification and characterisation of novel rare disease genes.
衣被蛋白复合体 1(COPI)在蛋白质和脂质从高尔基体到内质网(ER)的分拣和逆行运输中是不可或缺的。近年来,衣被蛋白已被牵连到被统称为“衣被蛋白病”的人类疾病中。
对两个具有神经发育综合征、可变小头畸形和白内障的家族进行全外显子或全基因组测序,揭示 COPB1 的双等位基因变异,该基因编码 COPI 的β-亚基(β-COP)。为了研究家族 1 的剪接供体位点变异,我们进行了患者血液 RNA 研究,并在 Xenopus tropicalis 基因组的同源区域使用 CRISPR/Cas9 对该变异进行建模。为了研究家族 2 的错义变异,我们研究了转染 COPB1 表达载体的人视网膜上皮细胞和胚胎肾细胞系的细胞表型,该载体引入了家族 2 的突变。
我们提出了一种新的隐性衣被蛋白病,其特征为严重的发育迟缓、白内障和可变的小头畸形。家族 1 的纯合剪接供体位点变异导致两种异常转录本,其中一种导致 COPB1 前 mRNA 中第 8 外显子的跳跃,以及 36 个氨基酸的框内缺失,导致 β-COP 和 β'-COP 之间的小相互作用界面的一个基序丢失。通过 CRISPR/Cas9 基因组编辑引入的同源突变的 Xenopus tropicalis 动物再现了人类综合征的特征,包括小头畸形和白内障。家族 2 的 COPB1 c.1651T>G p.Phe551Val 变异的体外建模确定了这种突变β-COP 的高尔基体到 ER 的回收循环缺陷,突变蛋白在高尔基体内被延迟。
这增加了越来越多的证据表明 COPI 亚基在大脑发育和人类健康中是必不可少的,并强调了外显子组和基因组测序与 Xenopus tropicalis CRISPR/Cas 建模相结合,用于鉴定和表征新型罕见疾病基因的实用性。