Heinrich Heine University, Medical Faculty, Institute for Medical Biometry and Bioinformatics, Moorenstraße 20, 40225 Düsseldorf, Germany.
Department of Genome Sciences, University of Washington School of Medicine, 3720 15th Avenue NE, Seattle, WA 98195-5065, USA.
Science. 2021 Apr 2;372(6537). doi: 10.1126/science.abf7117. Epub 2021 Feb 25.
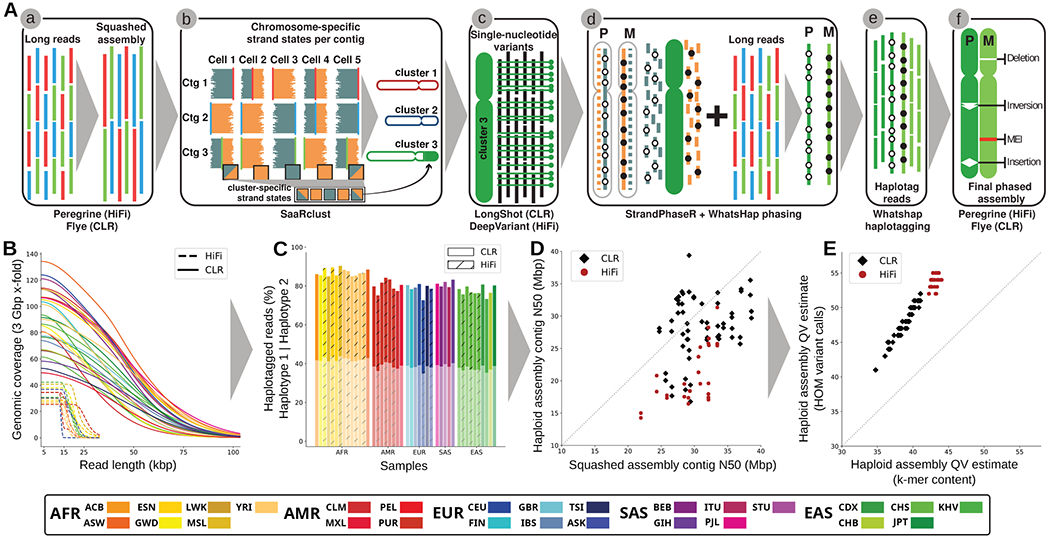
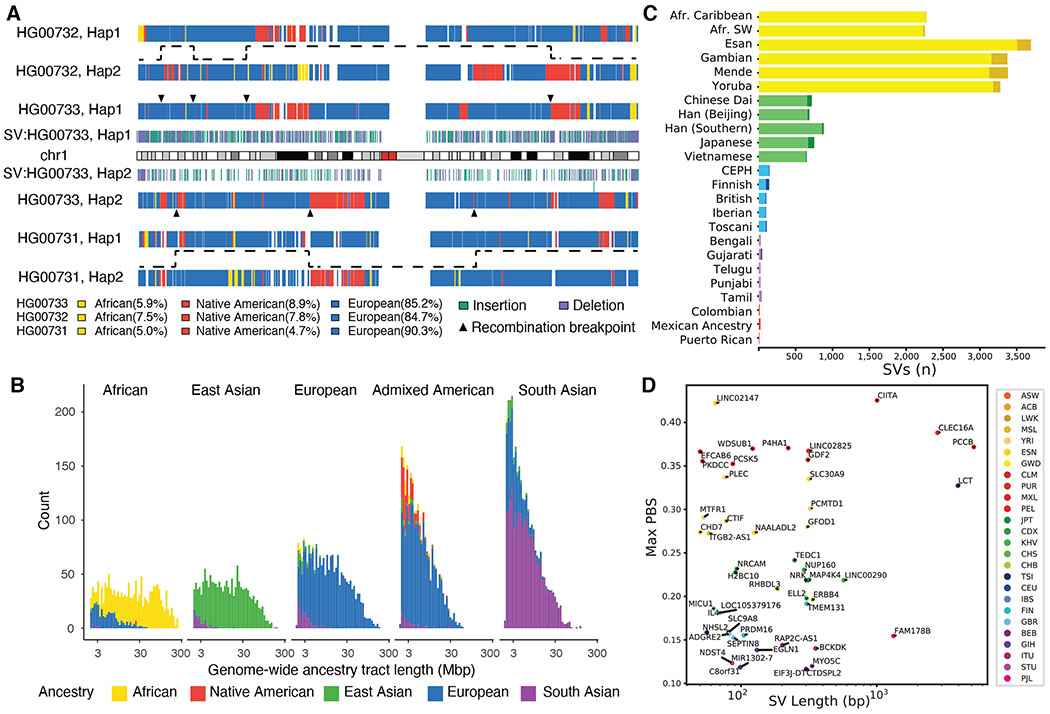
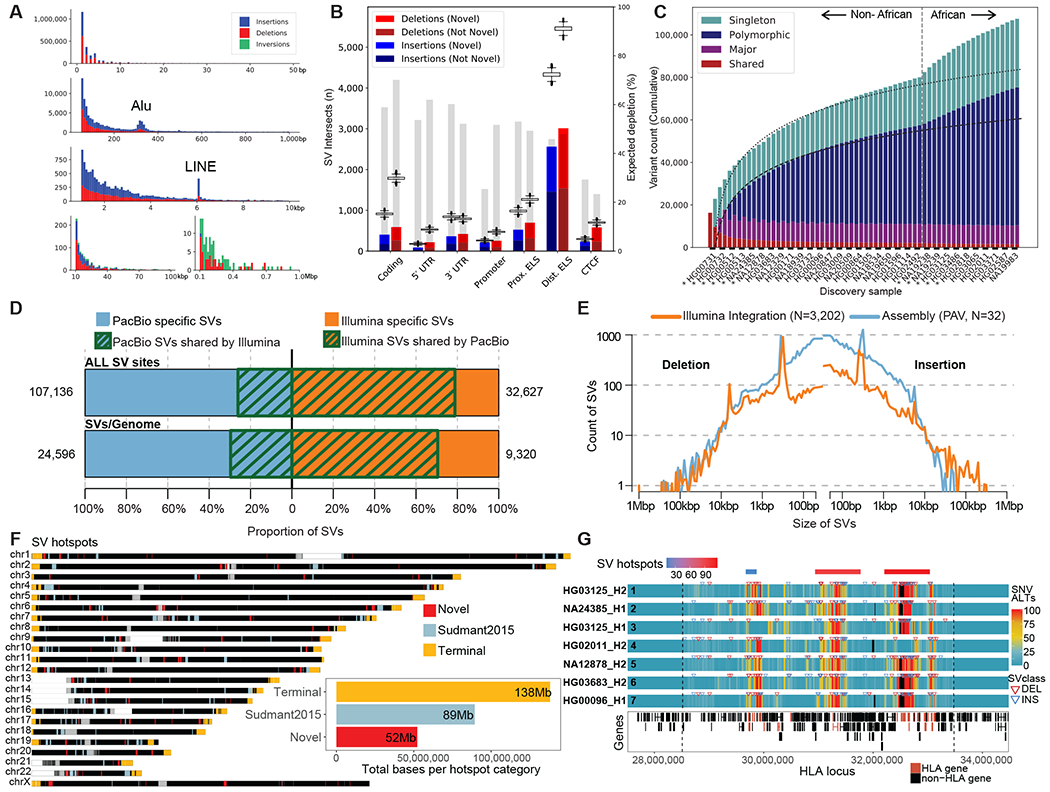
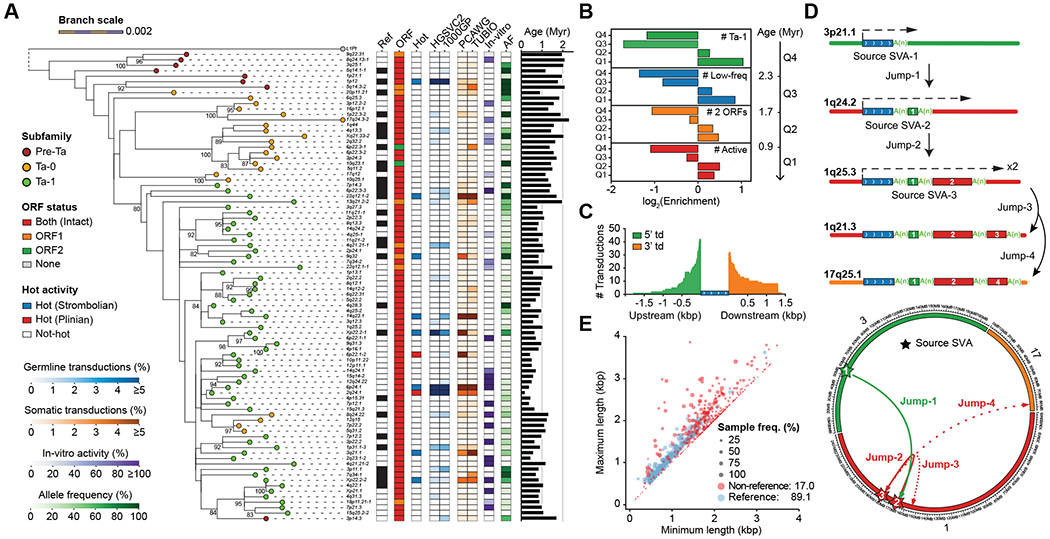
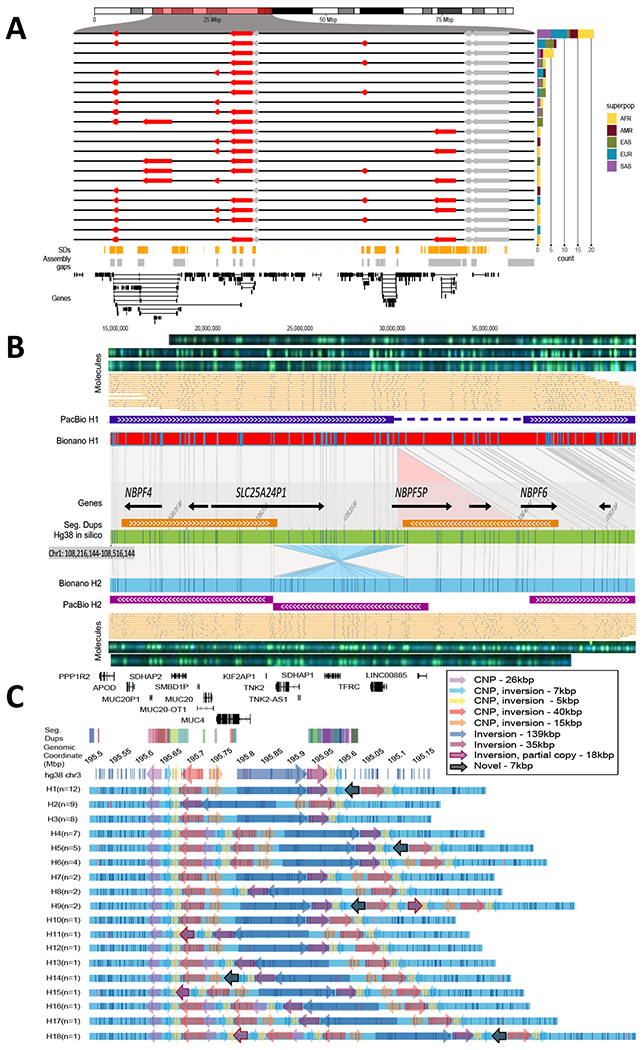
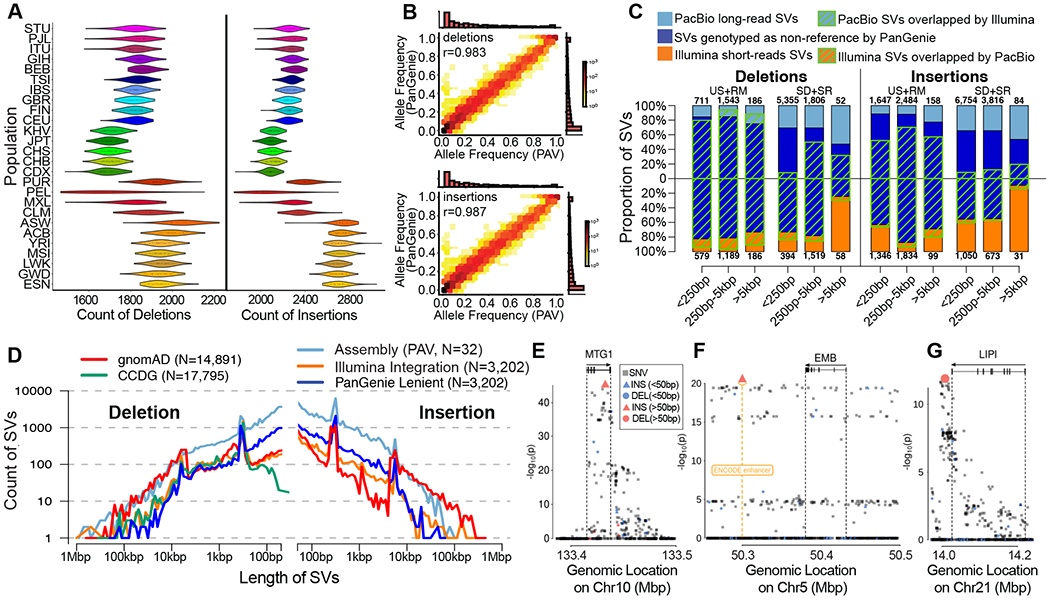
Long-read and strand-specific sequencing technologies together facilitate the de novo assembly of high-quality haplotype-resolved human genomes without parent-child trio data. We present 64 assembled haplotypes from 32 diverse human genomes. These highly contiguous haplotype assemblies (average minimum contig length needed to cover 50% of the genome: 26 million base pairs) integrate all forms of genetic variation, even across complex loci. We identified 107,590 structural variants (SVs), of which 68% were not discovered with short-read sequencing, and 278 SV hotspots (spanning megabases of gene-rich sequence). We characterized 130 of the most active mobile element source elements and found that 63% of all SVs arise through homology-mediated mechanisms. This resource enables reliable graph-based genotyping from short reads of up to 50,340 SVs, resulting in the identification of 1526 expression quantitative trait loci as well as SV candidates for adaptive selection within the human population.
长读长和单链测序技术结合在一起,可以在没有父母-子女三核苷酸数据的情况下从头组装高质量的单倍型解析人类基因组。我们展示了 32 个不同人类基因组中 64 个组装的单倍型。这些高度连续的单倍型组装体(覆盖基因组 50%所需的最小连续体长度:2600 万碱基对)整合了所有形式的遗传变异,甚至跨越复杂的基因座。我们鉴定了 107590 个结构变异(SVs),其中 68%是无法通过短读测序发现的,还有 278 个 SV 热点(跨越大片富含基因的序列)。我们对 130 个最活跃的移动元件源元件进行了特征分析,发现所有 SVs 中有 63%是通过同源介导机制产生的。该资源可以从多达 50340 个 SV 的短读中进行可靠的基于图的基因分型,从而鉴定出 1526 个表达数量性状基因座,以及人群中适应性选择的 SV 候选基因座。