Sympli Hakani D
Department of Biotechnology, School of Biosciences, Assam Don Bosco University, Tapesia, Kamarkuchi, Sonapur, Assam 782402 India.
Netw Model Anal Health Inform Bioinform. 2021;10(1):14. doi: 10.1007/s13721-020-00276-1. Epub 2021 Feb 24.
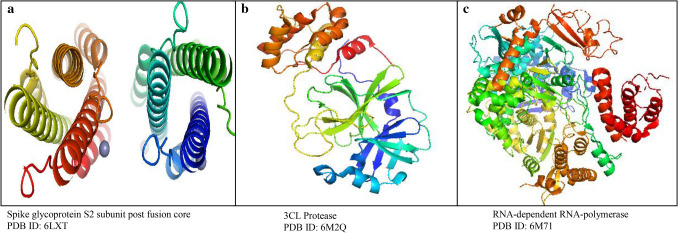
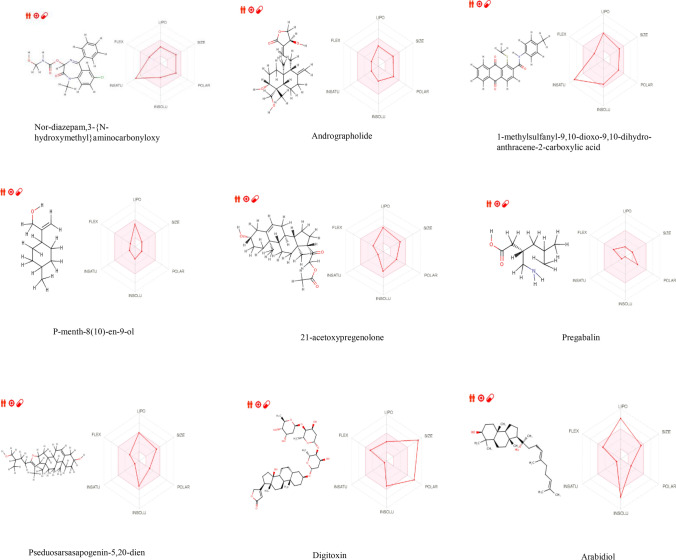
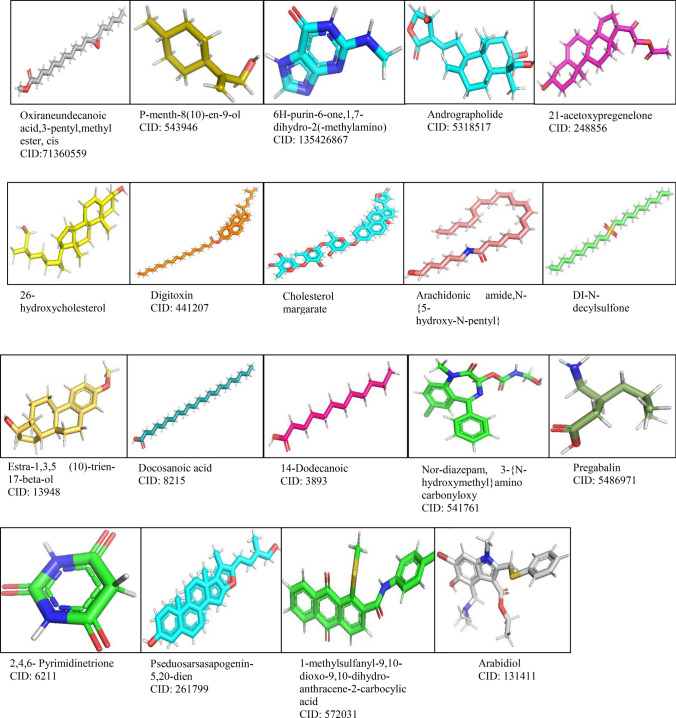
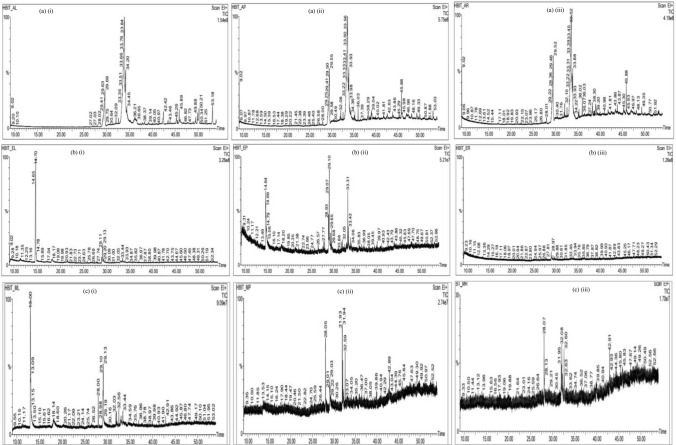
The main aim of the paper was to determine bioactive compounds in extracts using gas chromatographic technique and to investigate their drug-likeness potential using molecular docking algorithm and ADME studies on the recent intractable disease, for example, SARS-CoV-2. sample was prepared for GC-MS analysis. The peak components were identified based on the NIST Library. Molecular docking was performed using PatchDock, and energy refinement was carried out using the FireDock algorithm followed by drug-likeness analysis using the SwissADME tool. The mass spectrum revealed various pharmacologically important compounds and novel compounds 8-oxatetracyclo{5.2.1.1(2,6). 1(4,10)}dodecane, 7-tert-butyl-1,9,9-trimeth, docosane, 2,4-dimethyl, kryptogenin 2,4-dinitrophenyl hydrazine, and -decyl-alpha,-2-deoxyglycoside which are reported for the first time. Molecular docking using PatchDock illustrates GC-MS compounds Nor-diazepam,3-{-hydroxymethyl}aminocarbonyloxy a good docking and high binding affinity with atomic contact energy -10.95 kcal/mol against SARS-CoV-2 spike protein S2 subunit. ADME analysis predicts Nor-diazepam,3-{N-hydroxymethyl}aminocarbonyloxy and andrographolide showed very high drug-likeness parameters with no metabolism disturbances. The random control antiviral drug arabidiol revealed a lower binding affinity and lower solubility compared to bioactive compounds of . The study depicts the first and novel report on various pharmaceutical important GC-MS bioactive compounds and molecular docking study on having potential against various intractable diseases.
The online version contains supplementary material available at 10.1007/s13721-020-00276-1.
本文的主要目的是使用气相色谱技术测定提取物中的生物活性化合物,并使用分子对接算法以及针对近期难治性疾病(如SARS-CoV-2)的ADME研究来研究它们的类药潜力。制备样品用于GC-MS分析。基于NIST库鉴定峰成分。使用PatchDock进行分子对接,并使用FireDock算法进行能量优化,随后使用SwissADME工具进行类药分析。质谱显示了各种具有药理学重要性的化合物以及新型化合物8-氧杂四环{5.2.1.1(2,6).1(4,10)}十二烷、7-叔丁基-1,9,9-三甲基、二十二烷、2,4-二甲基、海可皂苷元2,4-二硝基苯肼和癸基-α,-2-脱氧糖苷,这些都是首次报道。使用PatchDock进行的分子对接表明,GC-MS化合物去甲西泮、3-{N-羟甲基}氨基羰基氧基与SARS-CoV-2刺突蛋白S2亚基具有良好的对接和高结合亲和力,原子接触能为-10.95 kcal/mol。ADME分析预测,去甲西泮、3-{N-羟甲基}氨基羰基氧基和穿心莲内酯显示出非常高的类药参数,且无代谢干扰。随机对照抗病毒药物阿拉伯二醇与……的生物活性化合物相比显示出较低的结合亲和力和较低的溶解度。该研究描述了关于各种具有药学重要性的GC-MS生物活性化合物的首个新颖报告以及对……具有抗各种难治性疾病潜力的分子对接研究。
在线版本包含可在10.1007/s13721-020-00276-1获取的补充材料。