Anh Duong T, Hai Pham-The, Huy Le D, Ngoc Hoang B, Ngoc Trinh T M, Dung Do T M, Park Eun J, Song In K, Kang Jong S, Kwon Joo-Hee, Tung Truong T, Han Sang-Bae, Nam Nguyen-Hai
Department of Pharmaceutical Chemistry, Hanoi University of Pharmacy, 13-15 Le Thanh Tong, Hanoi 10000, Vietnam.
College of Pharmacy, Chungbuk National University, 194-31, Osongsaengmyung-1, Heungdeok, Cheongju, Chungbuk 28160, Republic of Korea.
ACS Omega. 2021 Feb 8;6(7):4907-4920. doi: 10.1021/acsomega.0c05870. eCollection 2021 Feb 23.
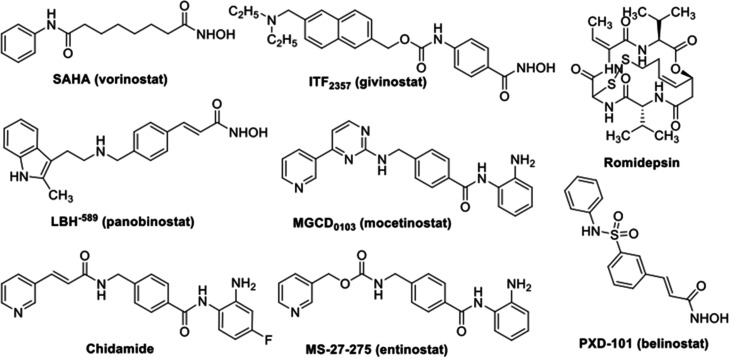
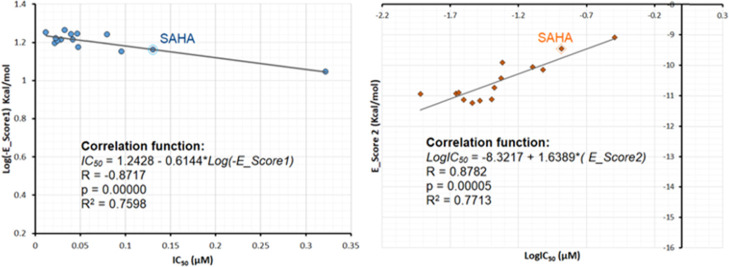
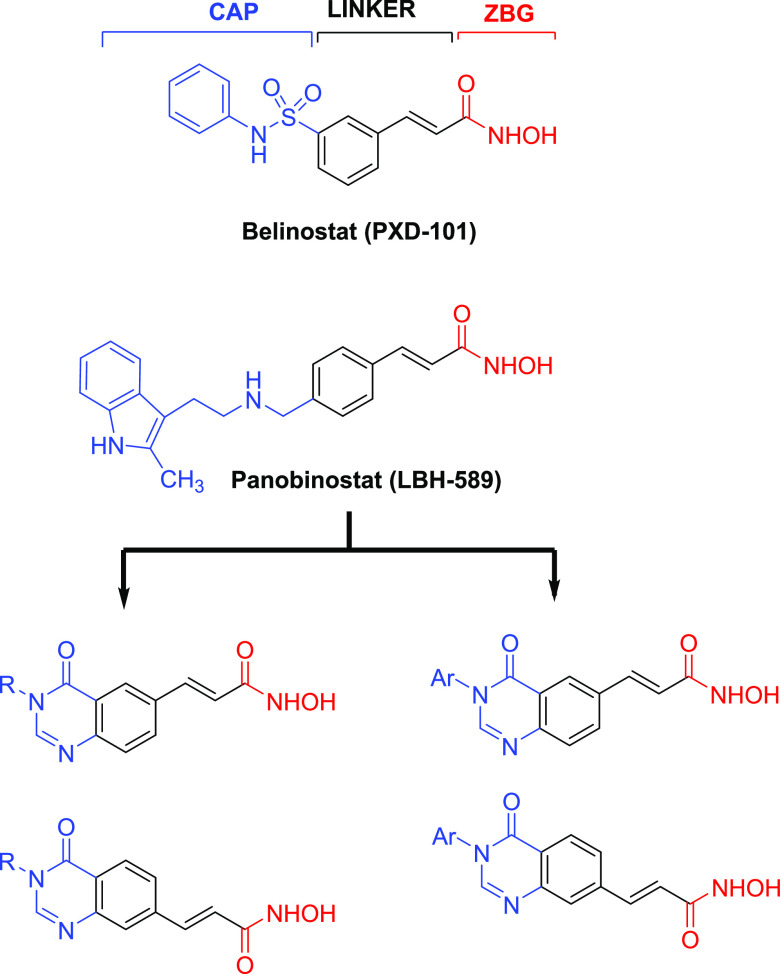
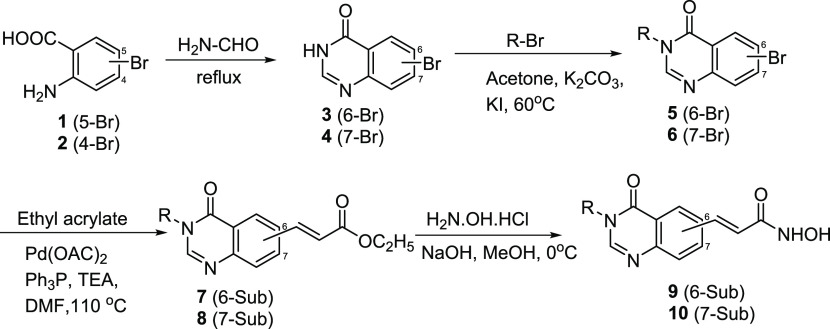
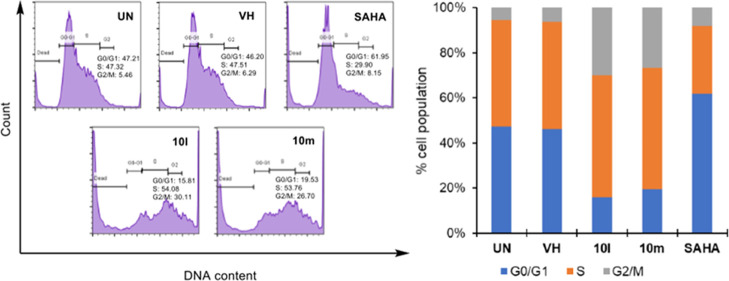
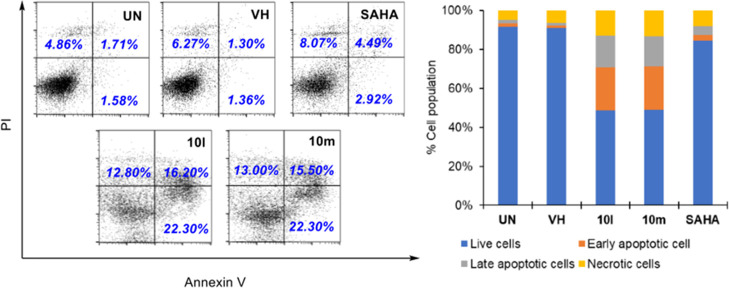
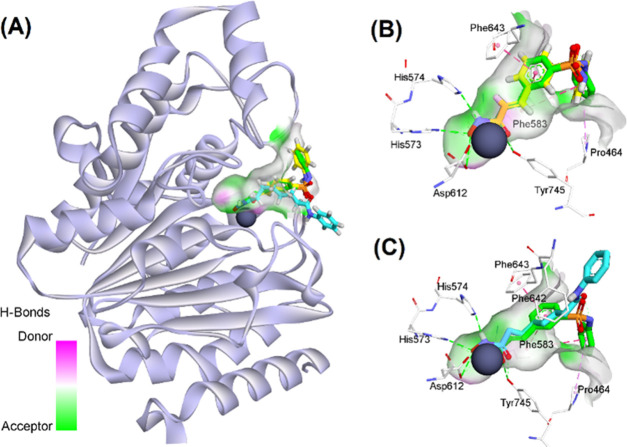
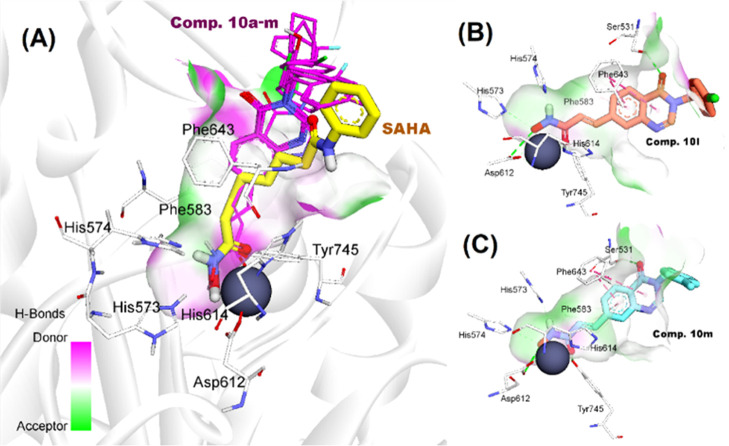
Two series of novel 4-oxoquinazoline-based -hydroxypropenamides (- and -) were designed, synthesized, and evaluated for their inhibitory and cytotoxicity activities against histone deacetylase (HDAC). The compounds showed good to potent HDAC inhibitory activity and cytotoxicity against three human cancer cell lines (SW620, colon; PC-3, prostate; NCI-H23, lung cancer). In this series, compounds with the -hydroxypropenamide functionality impeded at position 7 on the 4-oxoquinazoline skeleton (-) were generally more potent than compounds with the -hydroxypropenamide moiety at position 6 (-). Also, the -benzyl-substituted derivatives (-, -) exhibited stronger bioactivity than the -alkyl-substituted ones (-, -). Two compounds and were the most potent ones. Their HDAC inhibitory activity (IC values, 0.041-0.044 μM) and cytotoxicity (IC values, 0.671-1.211 μM) were approximately 2- to 3-fold more potent than suberoylanilide hydroxamic acid (SAHA). Some compounds showed up to 10-fold more potent HDAC6 inhibition compared to their inhibitory activity in total HDAC extract assay. Analysis of selected compounds and revealed that these compounds strongly induced both early and late apoptosis and arrested SW620 cells at the G2/M phase. Docking studies were carried out on the HDAC6 isoform for series - and revealed some important features contributing to the inhibitory activity of synthesized compounds.
设计、合成了基于4-氧代喹唑啉的两类新型β-羟基丙烯酰胺(α-和β-),并评估了它们对组蛋白脱乙酰酶(HDAC)的抑制活性和细胞毒性。这些化合物对三种人类癌细胞系(SW620,结肠癌;PC-3,前列腺癌;NCI-H23,肺癌)显示出良好至强效的HDAC抑制活性和细胞毒性。在该系列中,4-氧代喹唑啉骨架上7位带有β-羟基丙烯酰胺官能团的化合物(α-)通常比6位带有β-羟基丙烯酰胺部分的化合物(β-)更具活性。此外,苄基取代的衍生物(α-,β-)比烷基取代的衍生物(α-,β-)表现出更强的生物活性。两种化合物α和β是活性最强的。它们的HDAC抑制活性(IC值,0.041 - 0.044 μM)和细胞毒性(IC值,0.671 - 1.211 μM)比辛二酰苯胺异羟肟酸(SAHA)强约2至3倍。与它们在总HDAC提取物测定中的抑制活性相比,一些化合物对HDAC6的抑制作用高达10倍。对选定化合物α和β的分析表明,这些化合物强烈诱导早期和晚期凋亡,并使SW620细胞停滞在G2/M期。对α-系列的HDAC6亚型进行了对接研究,揭示了一些有助于合成化合物抑制活性的重要特征。