Guan Chenyang, Huang Xian, Yue Jinnan, Xiang Hongrui, Shaheen Samina, Jiang Zhenyan, Tao Yuexiao, Tu Jun, Liu Zhenshan, Yao Yufeng, Yang Wen, Hou Zhaoyuan, Liu Junling, Yang Xiao-Dong, Zou Qiang, Su Bing, Liu Zhiduo, Ni Jun, Cheng Jinke, Wu Xuefeng
Shanghai Institute of Immunology, Department of Immunology and Microbiology, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025 China.
Department of Biochemistry and Molecular Cell Biology, Shanghai Jiao Tong University School of Medicine, Shanghai, 200025 China.
Theranostics. 2021 Feb 15;11(8):3981-3995. doi: 10.7150/thno.55573. eCollection 2021.
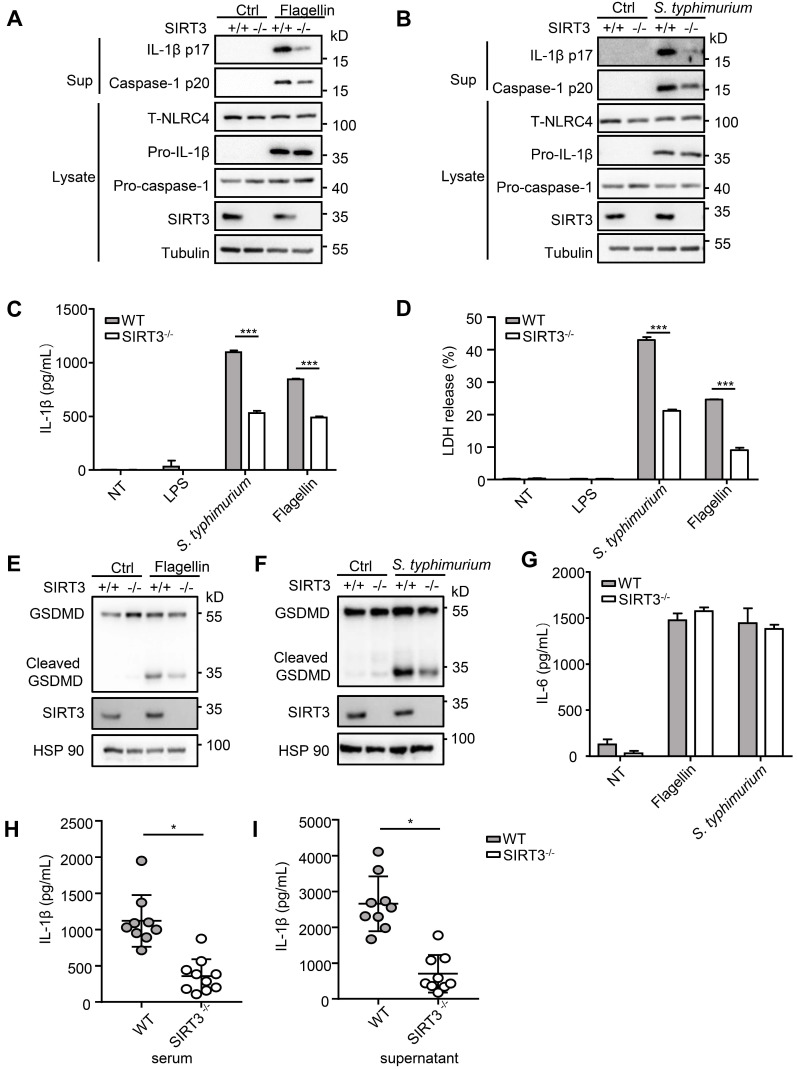
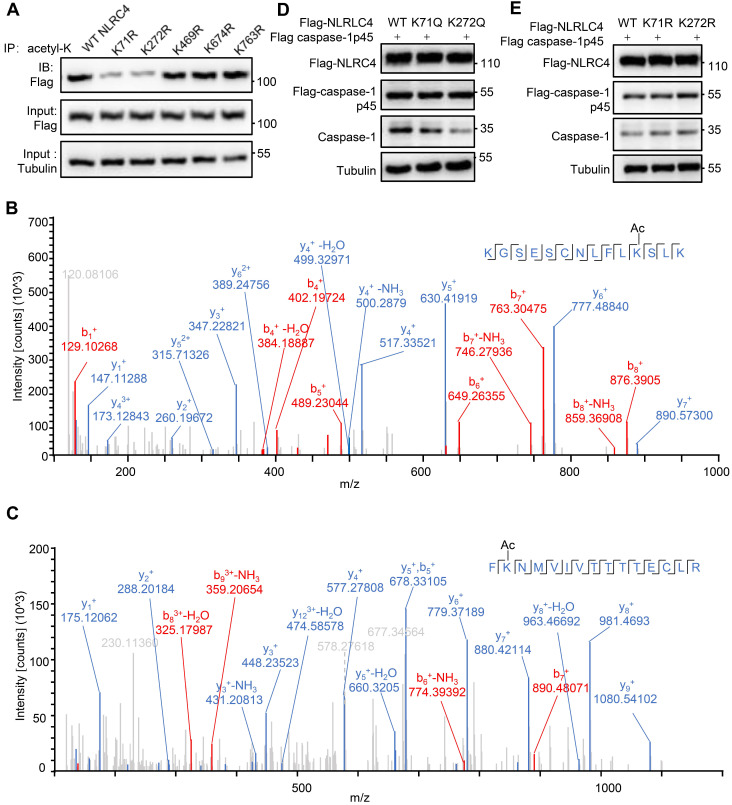
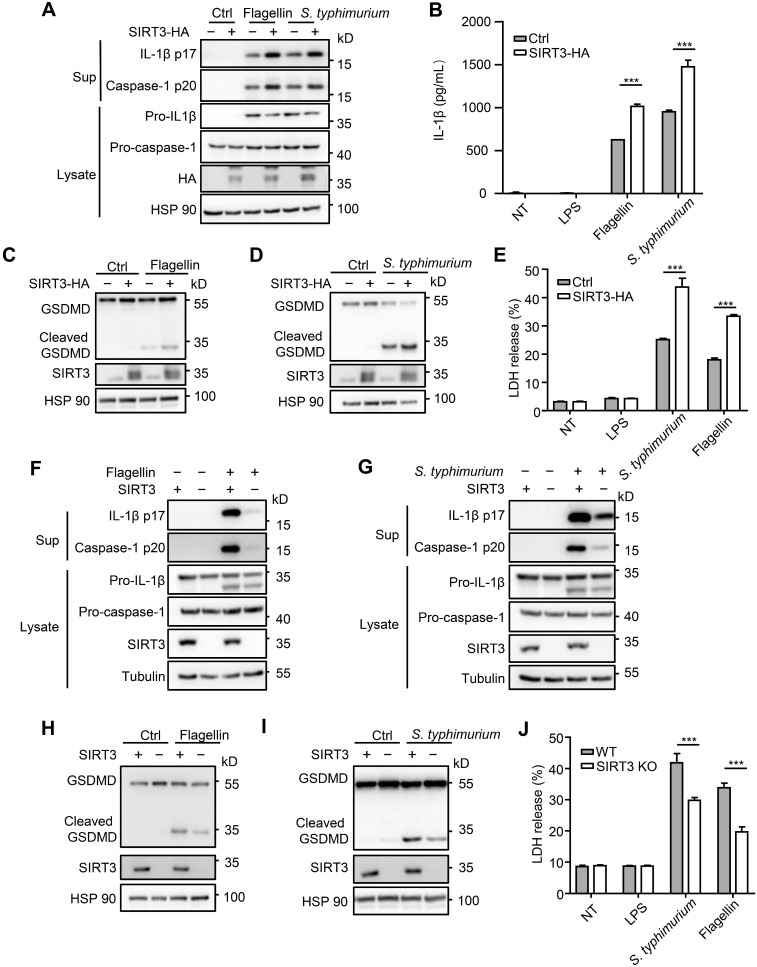
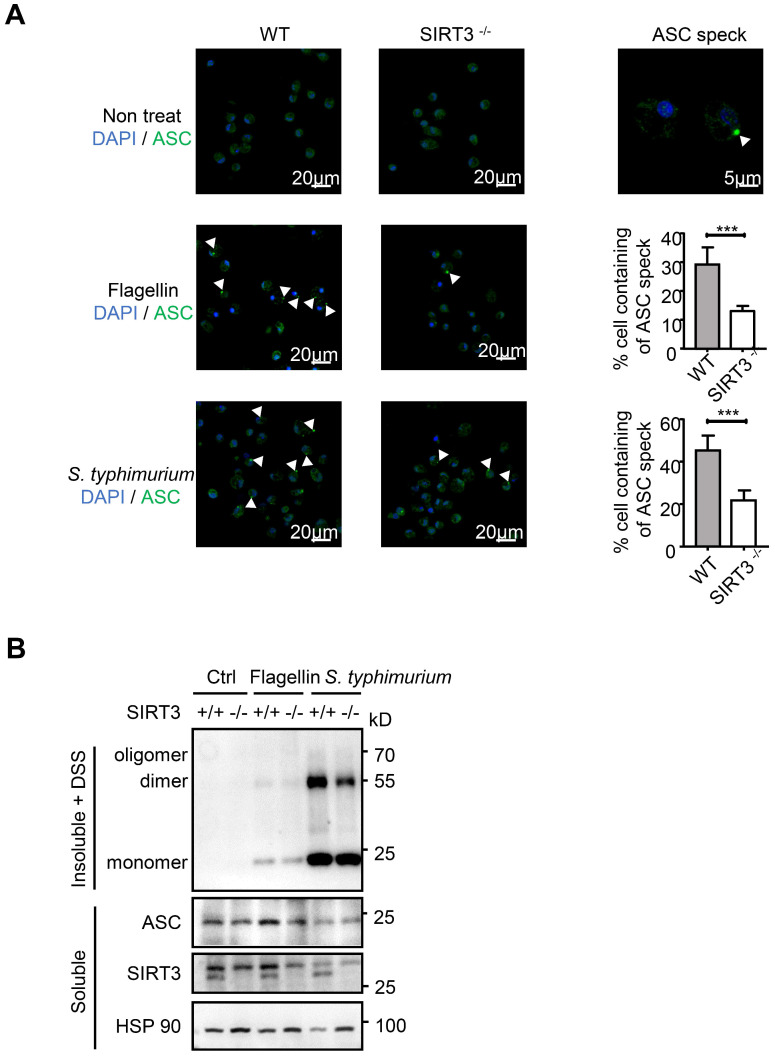
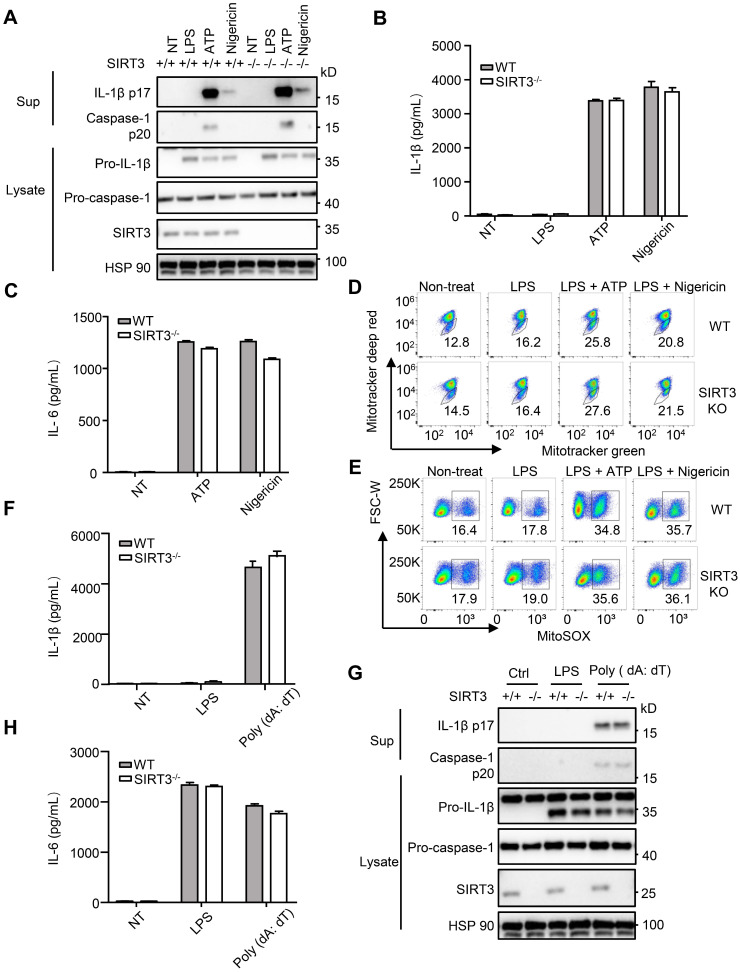
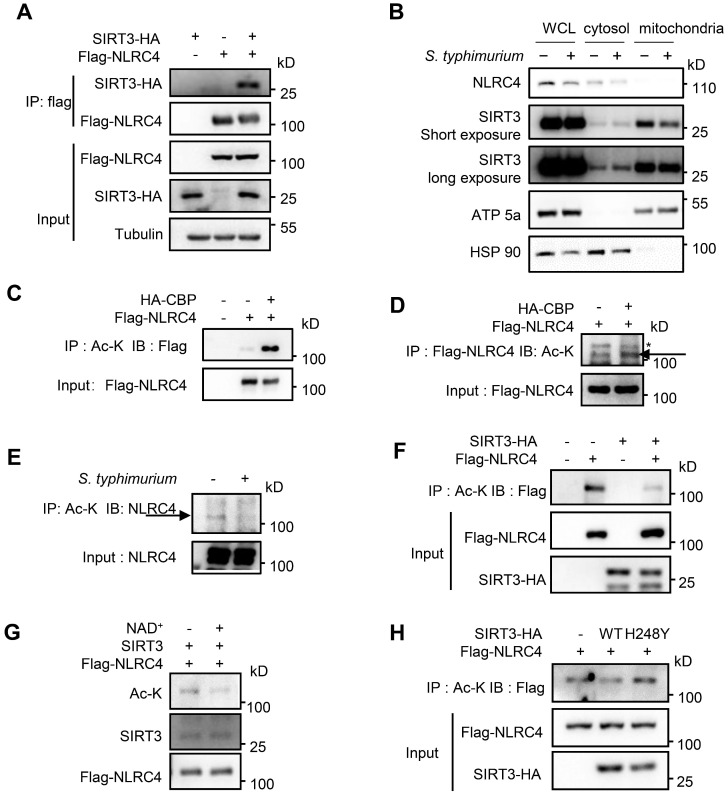
() infection of macrophage induces NLRC4 inflammasome-mediated production of the pro-inflammatory cytokines IL-1β. Post-translational modifications on NLRC4 are critical for its activation. Sirtuin3 (SIRT3) is the most thoroughly studied mitochondrial nicotinamide adenine dinucleotide (NAD) -dependent deacetylase. We wondered whether SIRT3 mediated-deacetylation could take part in NLRC4 inflammasome activation. We initially tested IL-1β production and pyroptosis after cytosolic transfection of flagellin or infection in wild type and SIRT3-deficient primary peritoneal macrophages via immunoblotting and ELISA assay. These results were confirmed in SIRT3-deficient immortalized bone marrow derived macrophages (iBMDMs) which were generated by CRISPR-Cas9 technology. In addition, experiments were conducted to confirm the role of SIRT3 in -induced cytokines production. Then NLRC4 assembly was analyzed by immune-fluorescence assay and ASC oligomerization assay. Immunoblotting, ELISA and flow cytometry were performed to clarify the role of SIRT3 in NLRP3 and AIM2 inflammasomes activation. To further investigate the mechanism of SIRT3 in NLRC4 activation, co-immunoprecipitation (Co-IP), we did immunoblot, cellular fractionation and in-vitro deacetylation assay. Finally, to clarify the acetylation sites of NLRC4, we performed liquid chromatography-mass spectrometry (LC-MS) and immunoblotting analysis. SIRT3 deficiency led to significantly impaired NLRC4 inflammasome activation and pyroptosis both and . Furthermore, SIRT3 promotes NLRC4 inflammasome assembly by inducing more ASC speck formation and ASC oligomerization. However, SIRT3 is dispensable for NLRP3 and AIM2 inflammasome activation. Moreover, SIRT3 interacts with and deacetylates NLRC4 to promote its activation. Finally, we proved that deacetylation of NLRC4 at Lys71 or Lys272 could promote its activation. Our study reveals that SIRT3 mediated-deacetylation of NLRC4 is pivotal for NLRC4 activation and the acetylation switch of NLRC4 may aid the clearance of infection.
(某种病原体)感染巨噬细胞会诱导NLRC4炎性小体介导促炎细胞因子IL-1β的产生。NLRC4的翻译后修饰对其激活至关重要。沉默调节蛋白3(SIRT3)是研究最深入的线粒体烟酰胺腺嘌呤二核苷酸(NAD)依赖性脱乙酰酶。我们想知道SIRT3介导的去乙酰化是否参与NLRC4炎性小体的激活。我们最初通过免疫印迹和ELISA检测,在野生型和SIRT3缺陷的原代腹腔巨噬细胞中进行鞭毛蛋白的胞质转染或(某种病原体)感染后,检测IL-1β的产生和细胞焦亡情况。这些结果在通过CRISPR-Cas9技术构建的SIRT3缺陷的永生化骨髓来源巨噬细胞(iBMDM)中得到了证实。此外,进行了实验以确认SIRT3在(某种病原体)诱导的细胞因子产生中的作用。然后通过免疫荧光检测和ASC寡聚化检测分析NLRC4的组装情况。进行免疫印迹、ELISA和流式细胞术以阐明SIRT3在NLRP3和AIM2炎性小体激活中的作用。为了进一步研究SIRT3在NLRC4激活中的机制,我们进行了免疫共沉淀(Co-IP)、免疫印迹、细胞分级分离和体外去乙酰化检测。最后,为了阐明NLRC4的乙酰化位点,我们进行了液相色谱-质谱联用(LC-MS)和免疫印迹分析。SIRT3缺陷导致(某种病原体)感染时NLRC4炎性小体激活和细胞焦亡均显著受损。此外,SIRT3通过诱导更多的ASC斑点形成和ASC寡聚化来促进NLRC4炎性小体的组装。然而,SIRT3对于NLRP3和AIM2炎性小体的激活是可有可无的。而且,SIRT3与NLRC4相互作用并使其去乙酰化以促进其激活。最后,我们证明NLRC4在赖氨酸71或赖氨酸272处的去乙酰化可促进其激活。我们的研究表明,SIRT3介导的NLRC4去乙酰化对于NLRC4激活至关重要,NLRC4的乙酰化开关可能有助于清除(某种病原体)感染。