Center of Experimental Rheumatology, Department of Rheumatology, University Hospital Zurich, University of Zurich, 8952 Schlieren, Switzerland.
Institute of Immunobiology, Cantonal Hospital St. Gallen, 9007 St. Gallen, Switzerland.
Int J Mol Sci. 2021 Feb 13;22(4):1861. doi: 10.3390/ijms22041861.
Pathological activation of cardiac fibroblasts is a key step in development and progression of cardiac fibrosis and heart failure. This process has been associated with enhanced autophagocytosis, but molecular mechanisms remain largely unknown.
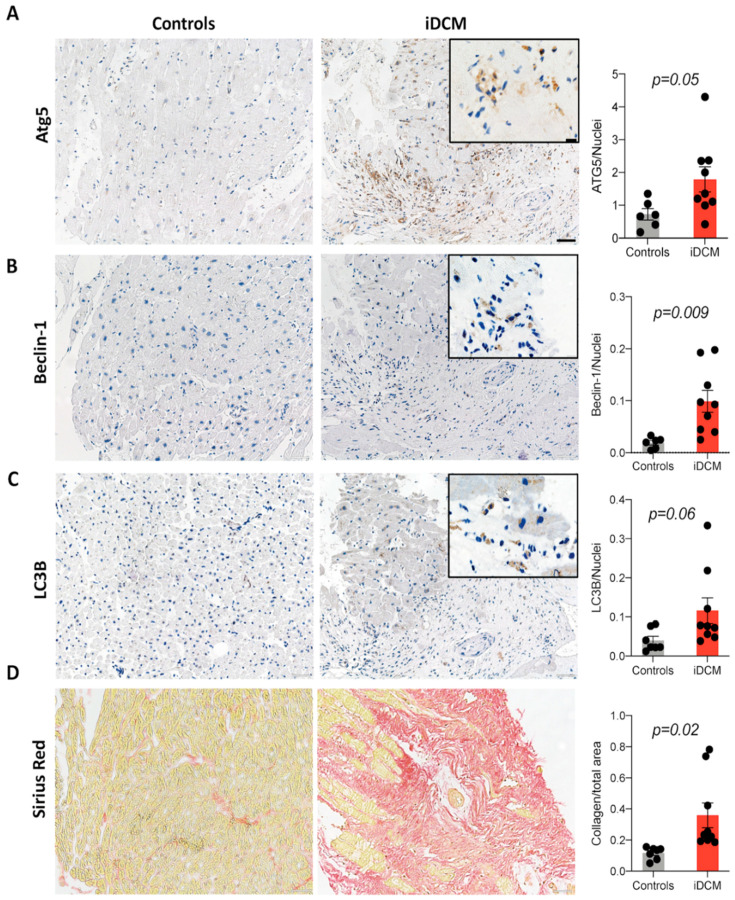
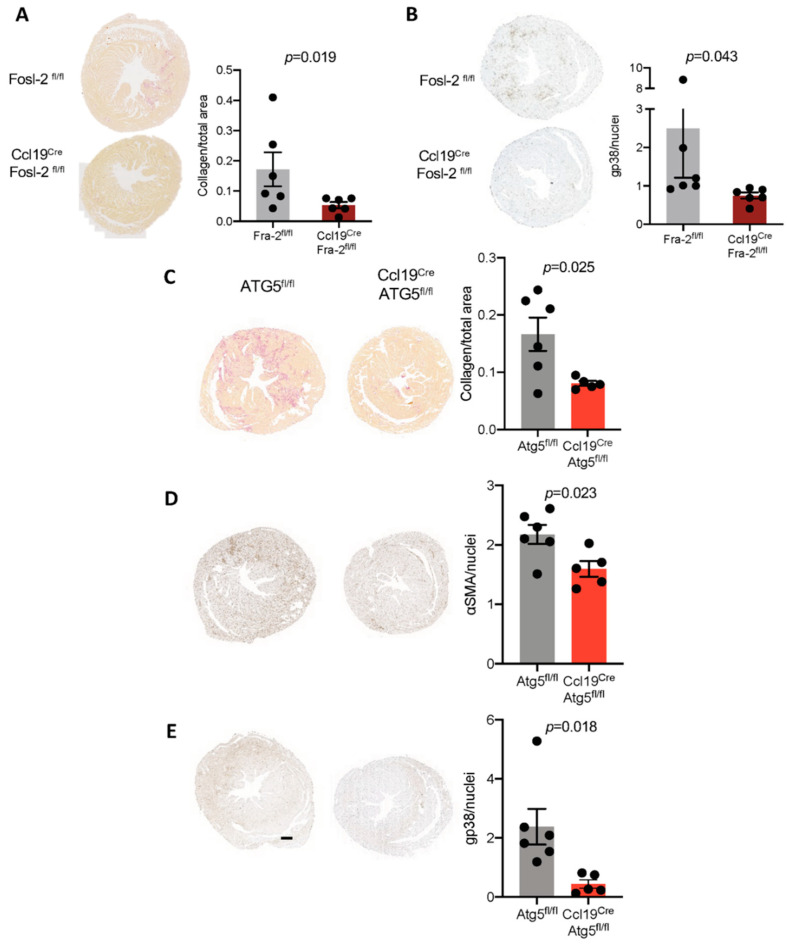
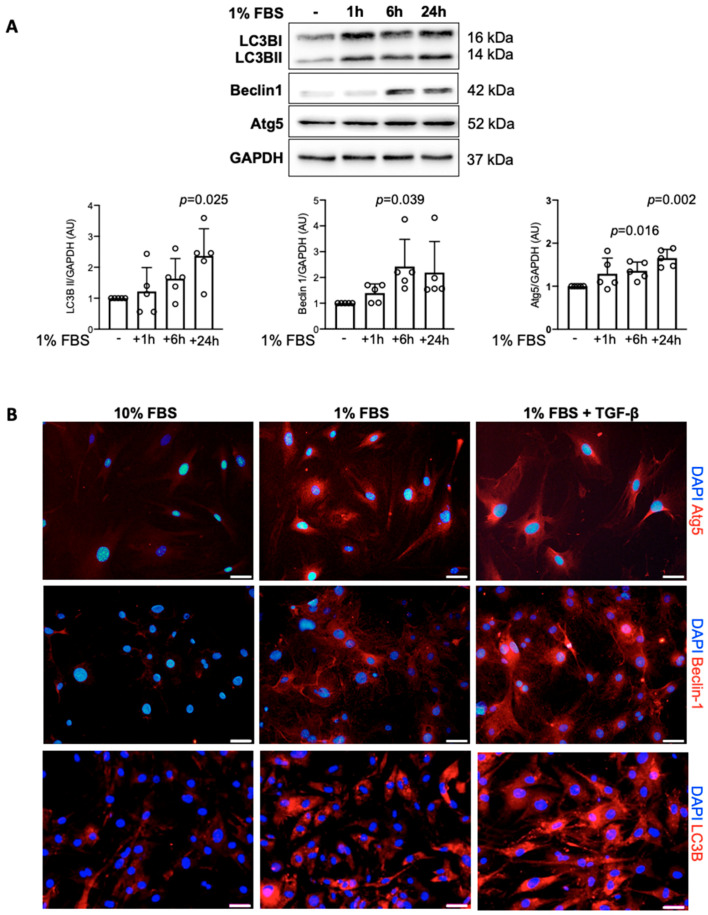
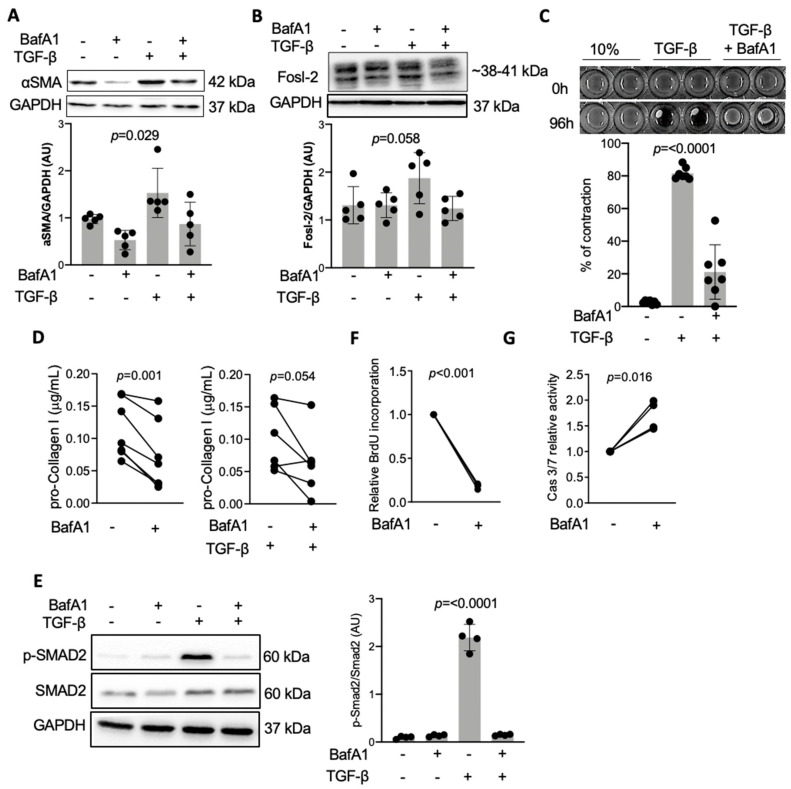
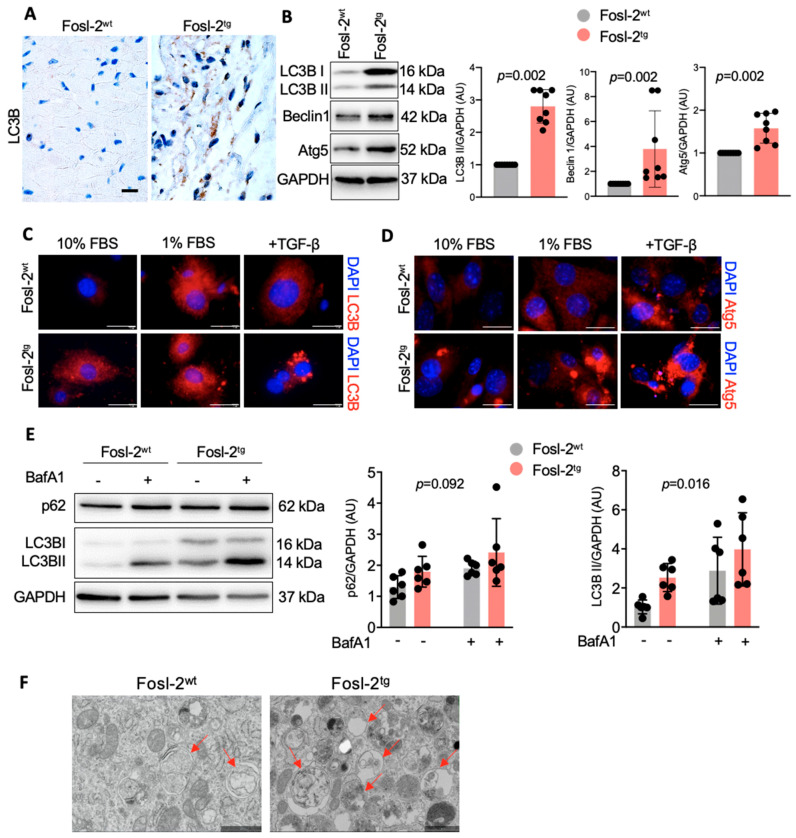
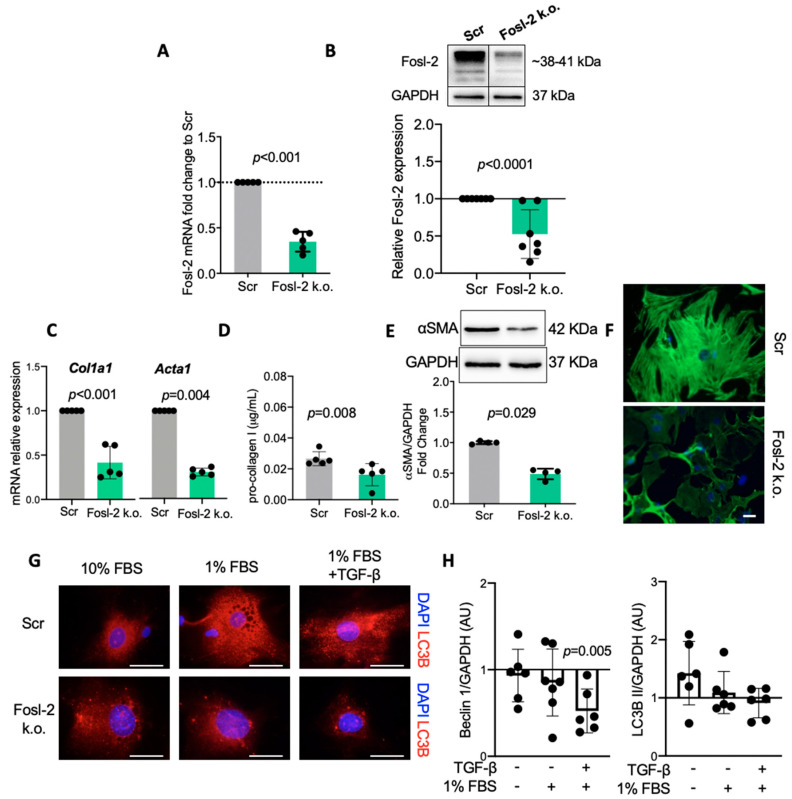
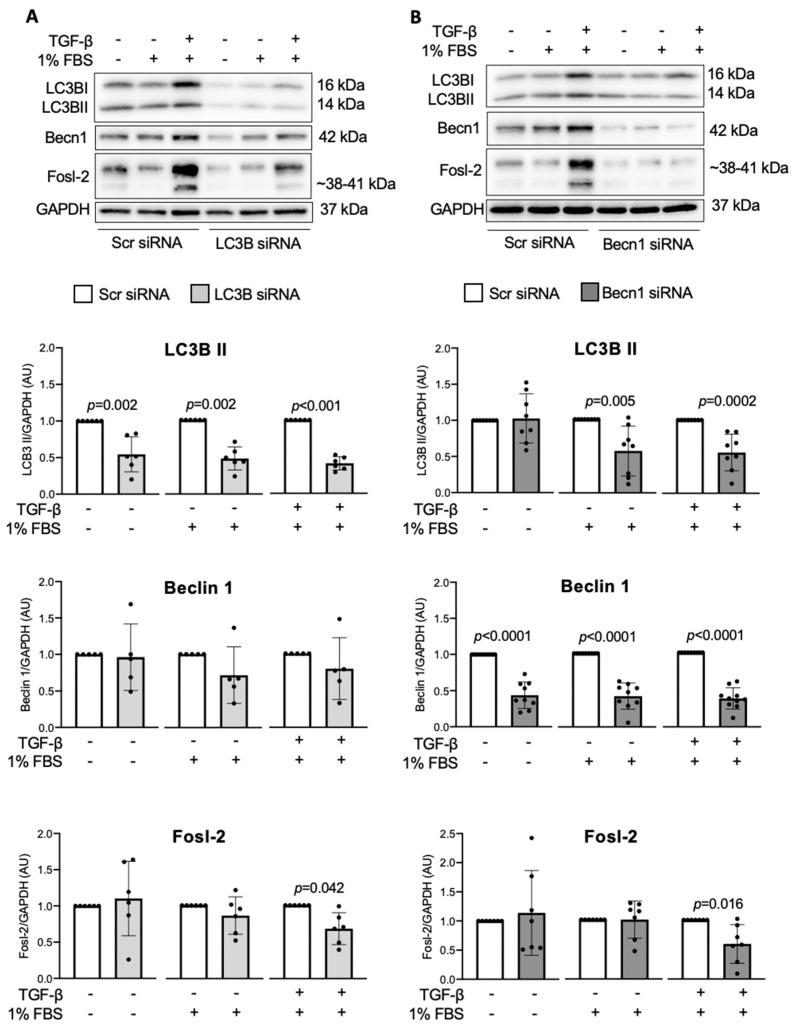
Immunohistochemical analysis of endomyocardial biopsies showed increased activation of autophagy in fibrotic hearts of patients with inflammatory cardiomyopathy. In vitro experiments using mouse and human cardiac fibroblasts confirmed that blockade of autophagy with Bafilomycin A1 inhibited fibroblast-to-myofibroblast transition induced by transforming growth factor (TGF)-β. Next, we observed that cardiac fibroblasts obtained from mice overexpressing transcription factor Fos-related antigen 2 (Fosl-2tg) expressed elevated protein levels of autophagy markers: the lipid modified form of microtubule-associated protein 1A/1B-light chain 3B (LC3BII), Beclin-1 and autophagy related 5 (Atg5). In complementary experiments, silencing of Fosl-2 with antisense GapmeR oligonucleotides suppressed production of type I collagen, myofibroblast marker alpha smooth muscle actin and autophagy marker Beclin-1 in cardiac fibroblasts. On the other hand, silencing of either LC3B or Beclin-1 reduced Fosl-2 levels in TGF-β-activated, but not in unstimulated cells. Using a cardiac hypertrophy model induced by continuous infusion of angiotensin II with osmotic minipumps, we confirmed that mice lacking either Fosl-2 (Ccl19CreFosl2flox/flox) or Atg5 (Ccl19CreAtg5flox/flox) in stromal cells were protected from cardiac fibrosis.
Our findings demonstrate that Fosl-2 regulates autophagocytosis and the TGF-β-Fosl-2-autophagy axis controls differentiation of cardiac fibroblasts. These data provide a new insight for the development of pharmaceutical targets in cardiac fibrosis.
心肌成纤维细胞的病理性激活是心肌纤维化和心力衰竭发展和进展的关键步骤。这一过程与自噬作用增强有关,但分子机制仍知之甚少。
对心肌活检的免疫组织化学分析显示,炎症性心肌病患者纤维化心脏中的自噬活性增加。使用小鼠和人心脏成纤维细胞的体外实验证实,用巴弗洛霉素 A1 阻断自噬可抑制转化生长因子(TGF)-β诱导的成纤维细胞向肌成纤维细胞的转化。接下来,我们观察到过表达转录因子 Fos 相关抗原 2(Fosl-2tg)的小鼠心脏成纤维细胞表达了升高的自噬标志物:微管相关蛋白 1A/1B-轻链 3B(LC3BII)、Beclin-1 和自噬相关蛋白 5(Atg5)的脂质修饰形式。在补充实验中,用反义 GapmeR 寡核苷酸沉默 Fosl-2 可抑制心脏成纤维细胞中 I 型胶原、平滑肌肌动蛋白和自噬标志物 Beclin-1 的产生。另一方面,沉默 LC3B 或 Beclin-1 可降低 TGF-β激活但未刺激细胞中 Fosl-2 的水平。通过使用持续输注血管紧张素 II 的渗透微型泵诱导的心肌肥厚模型,我们证实基质细胞中缺乏 Fosl-2(Ccl19CreFosl2flox/flox)或 Atg5(Ccl19CreAtg5flox/flox)的小鼠可防止心肌纤维化。
我们的研究结果表明,Fosl-2 调节自噬作用,TGF-β-Fosl-2-自噬轴控制心脏成纤维细胞的分化。这些数据为心脏纤维化的药物靶点开发提供了新的见解。