Dabaj Ivana, Sudrié-Arnaud Bénédicte, Lecoquierre François, Raymond Kimiyo, Ducatez Franklin, Guerrot Anne-Marie, Snanoudj Sarah, Coutant Sophie, Saugier-Veber Pascale, Marret Stéphane, Nicolas Gaël, Tebani Abdellah, Bekri Soumeya
Department of Neonatal Pediatrics, Intensive Care and Neuropediatrics, Normandie University, UNIROUEN, CHU Rouen, INSERM U1245, 76000 Rouen, France.
Department of Metabolic Biochemistry, Normandie University, UNIROUEN, CHU Rouen, INSERM U1245, 76000 Rouen, France.
Life (Basel). 2021 Feb 27;11(3):187. doi: 10.3390/life11030187.
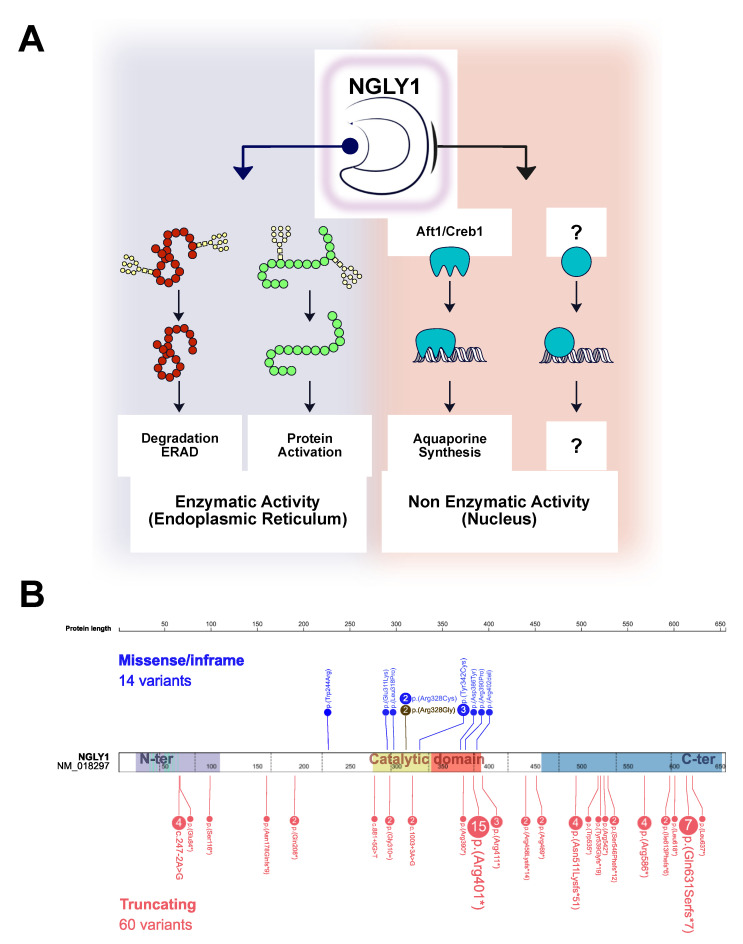
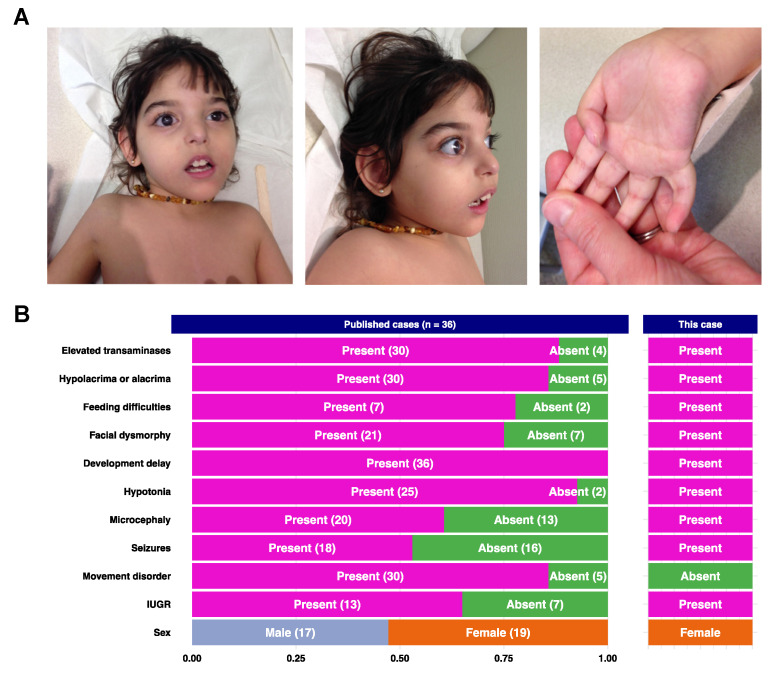
NGLY1 deficiency is the first recognized autosomal recessive disorder of N-linked deglycosylation (NGLY1-CDDG). This severe multisystemic disease is still poorly known and, to date, most cases have been diagnosed through whole exome or genome sequencing. The aim of this study is to provide the clinical, biochemical and molecular description of the first NGLY1-CDDG patient from France along with a literature review. The index case presented with developmental delay, acquired microcephaly, hypotonia, alacrimia, feeding difficulty, and dysmorphic features. Given the complex clinical picture and the multisystemic involvement, a trio-based exome sequencing was conducted and urine oligosaccharides were assessed using mass spectrometry. The exome sequencing revealed a novel variant in the gene in a homozygous state. NGLY1 deficiency was confirmed by the identification of the Neu5Ac1Hex1GlcNAc1-Asn oligosaccharide in the urine of the patient. Literature review revealed the association of some key clinical and biological features such as global developmental delay-hypertransaminasemia, movement disorders, feeding difficulties and alacrima/hypolacrima.
NGLY1缺乏症是首个被确认的N-连接去糖基化常染色体隐性疾病(NGLY1-CDDG)。这种严重的多系统疾病仍鲜为人知,迄今为止,大多数病例是通过全外显子组或基因组测序确诊的。本研究旨在对法国首例NGLY1-CDDG患者进行临床、生化和分子描述,并进行文献综述。索引病例表现为发育迟缓、后天性小头畸形、肌张力减退、无泪、喂养困难和畸形特征。鉴于复杂的临床症状和多系统受累情况,进行了基于三联体的外显子组测序,并使用质谱法评估尿寡糖。外显子组测序在该基因中发现了一个纯合状态的新变异。通过在患者尿液中鉴定出Neu5Ac1Hex1GlcNAc1-Asn寡糖,证实了NGLY1缺乏症。文献综述揭示了一些关键临床和生物学特征之间的关联,如全面发育迟缓-高转氨酶血症、运动障碍、喂养困难和无泪/泪液分泌减少。