Koo Bon-Kyung, Munroe William, Gralla Edith B, Valentine Joan Selverstone, Whitelegge Julian P
Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, CA, United States.
The Pasarow Mass Spectrometry Laboratory, David Geffen School of Medicine, NPI-Semel Institute for Neuroscience and Human Behavior, University of California, Los Angeles, Los Angeles, CA, United States.
Front Neurosci. 2021 Feb 18;14:619279. doi: 10.3389/fnins.2020.619279. eCollection 2020.
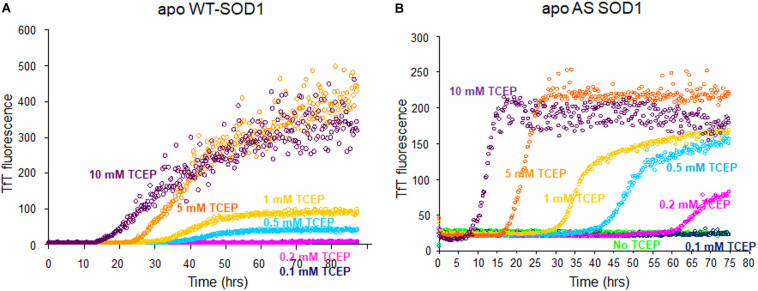
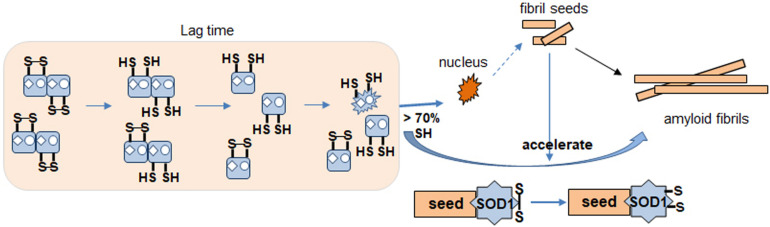
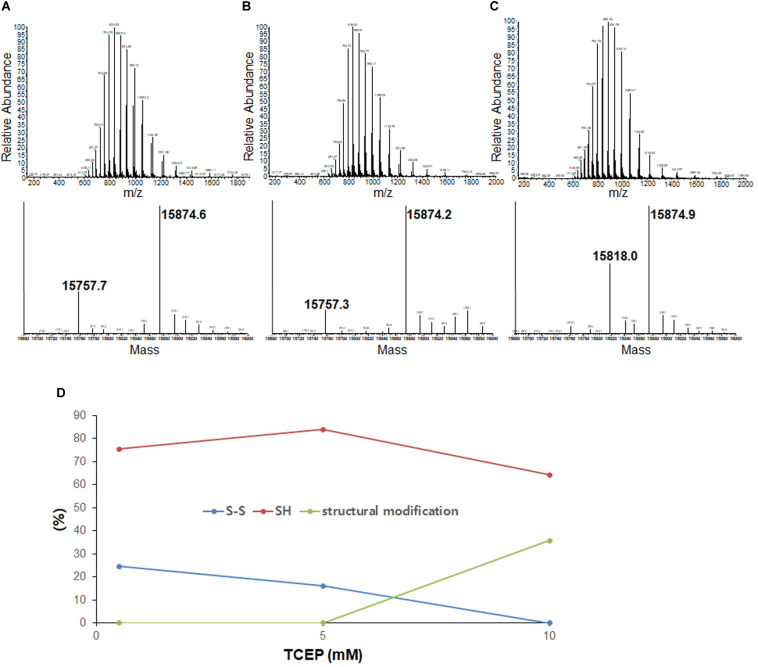
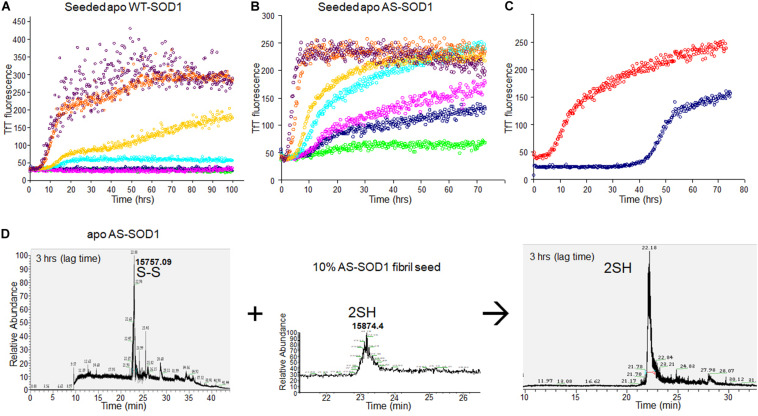
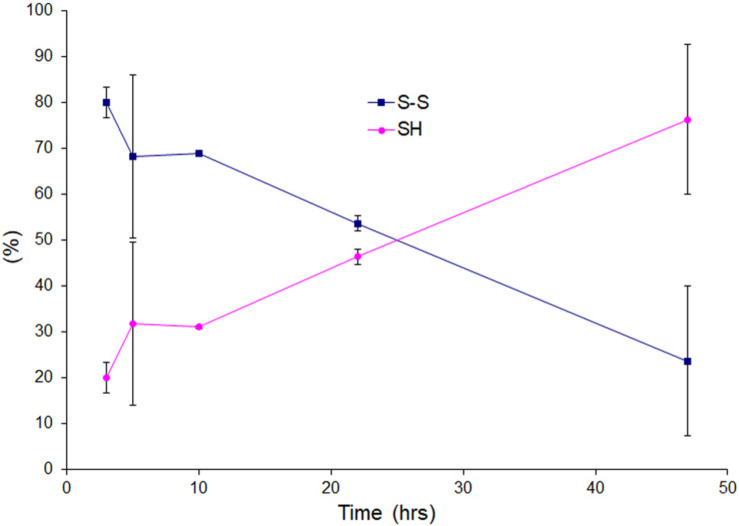
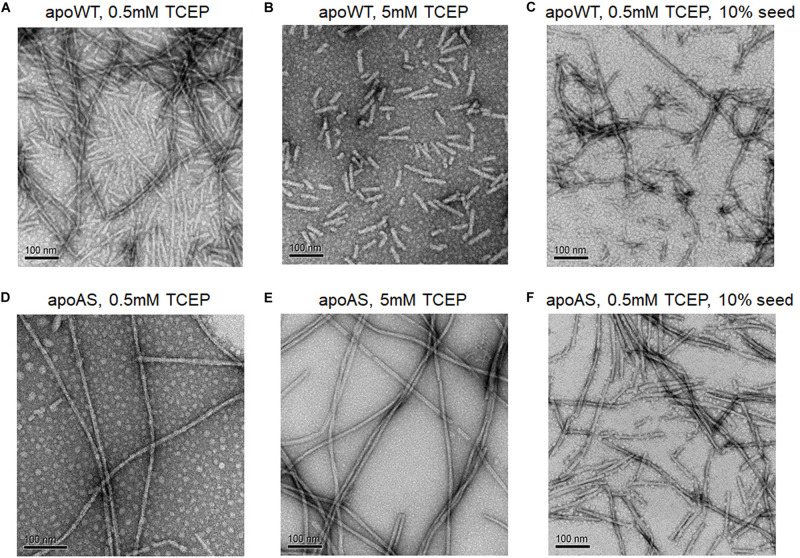
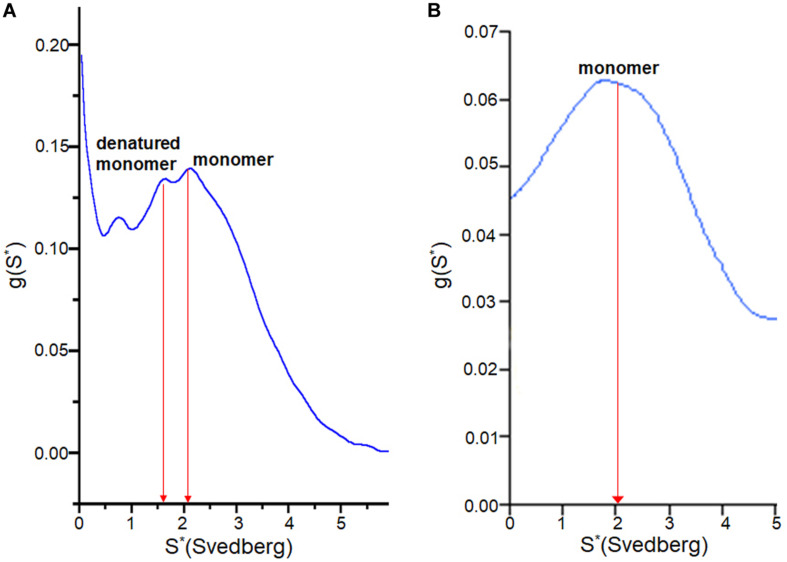
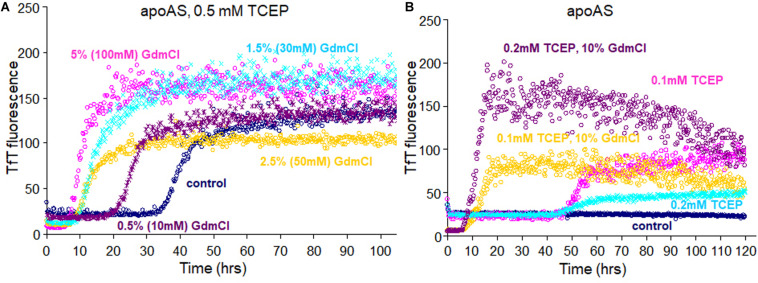
Wild-type human SOD1 forms a highly conserved intra-molecular disulfide bond between C57-C146, and in its native state is greatly stabilized by binding one copper and one zinc atom per monomer rendering the protein dimeric. Loss of copper extinguishes dismutase activity and destabilizes the protein, increasing accessibility of the disulfide with monomerization accompanying disulfide reduction. A further pair of free thiols exist at C6 and C111 distant from metal binding sites, raising the question of their function. Here we investigate their role in misfolding of SOD1 along a pathway that leads to formation of amyloid fibrils. We present the seeding reaction of a mutant SOD1 lacking free sulfhydryl groups (AS-SOD1) to exclude variables caused by these free cysteines. Completely reduced fibril seeds decreasing the kinetic barrier to cleave the highly conserved intramolecular disulfide bond, and accelerating SOD1 reduction and initiation of fibrillation. Presence or absence of the pair of free thiols affects kinetics of fibrillation. Previously, we showed full maturation with both Cu and Zn prevents this behavior while lack of Cu renders sensitivity to fibrillation, with presence of the native disulfide bond modulating this propensity much more strongly than presence of Zn or dimerization. Here we further investigate the role of reduction of the native C57-C146 disulfide bond in fibrillation of wild-type hSOD1, firstly through removal of free thiols by paired mutations C6A, C111S (AS-SOD1), and secondly in seeded fibrillation reactions modulated by reductant tris (2-carboxyethyl) phosphine (TCEP). Fibrillation of AS-SOD1 was dependent upon disulfide reduction and showed classic lag and exponential growth phases compared with wild-type hSOD1 whose fibrillation trajectories were typically somewhat perturbed. Electron microscopy showed that AS-SOD1 formed classic fibrils while wild-type fibrillation reactions showed the presence of smaller "sausage-like" oligomers in addition to fibrils, highlighting the potential for mixed disulfides involving C6/C111 to disrupt efficient fibrillation. Seeding by addition of sonicated fibrils lowered the TCEP concentration needed for fibrillation in both wild-type and AS-SOD1 providing evidence for template-driven structural disturbance that elevated susceptibility to reduction and thus propensity to fibrillate.
野生型人类超氧化物歧化酶1(SOD1)在C57 - C146之间形成高度保守的分子内二硫键,在其天然状态下,每个单体通过结合一个铜原子和一个锌原子而得到极大稳定,使该蛋白质形成二聚体。铜的缺失会消除歧化酶活性并使蛋白质不稳定,随着二硫键还原伴随单体化,二硫键的可及性增加。在远离金属结合位点的C6和C111处还存在另一对游离巯基,这就引发了它们功能的问题。在这里,我们沿着导致淀粉样纤维形成的途径研究它们在SOD1错误折叠中的作用。我们展示了缺乏游离巯基的突变型SOD1(AS - SOD1)的种子反应,以排除由这些游离半胱氨酸引起的变量。完全还原的纤维种子降低了切割高度保守的分子内二硫键的动力学障碍,并加速了SOD1的还原和纤维形成的起始。这对游离巯基的存在与否会影响纤维形成的动力学。此前,我们表明同时存在铜和锌时SOD1完全成熟可防止这种行为,而缺乏铜则使其对纤维形成敏感,天然二硫键的存在比锌的存在或二聚化更强烈地调节这种倾向。在这里,我们进一步研究野生型hSOD1中天然C57 - C146二硫键还原在纤维形成中的作用,首先通过C6A、C111S双突变(AS - SOD1)去除游离巯基,其次在由还原剂三(2 - 羧乙基)膦(TCEP)调节种子纤维形成反应中进行研究。与野生型hSOD1相比,AS - SOD1的纤维形成依赖于二硫键还原,并表现出典型的延迟期和指数生长期,野生型hSOD1的纤维形成轨迹通常会受到一定干扰。电子显微镜显示,AS - SOD1形成典型的纤维,而野生型纤维形成反应除了纤维外还显示存在较小的“香肠状”寡聚体,突出了涉及C6/C111的混合二硫键破坏有效纤维形成的可能性。通过添加超声处理的纤维进行种子接种降低了野生型和AS - SOD1纤维形成所需的TCEP浓度,为模板驱动的结构干扰提供了证据,这种干扰提高了对还原的敏感性,从而增加了纤维形成的倾向。