Shandong Provincial Key Laboratory of Biophysics (Dezhou University), Dezhou, Shandong 253023, China.
Department of Physics, Dezhou University, Dezhou, Shandong 253023, China.
Technol Health Care. 2021;29(S1):103-114. doi: 10.3233/THC-218011.
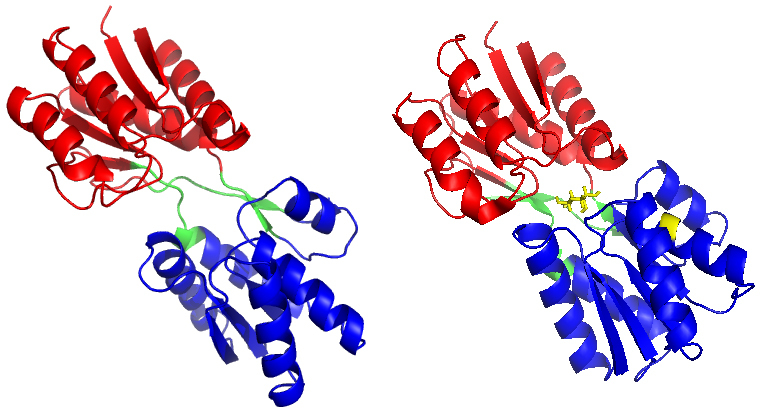
The ribose-binding protein (RBP) from Escherichia coli is one of the representative structures of periplasmic binding proteins. Binding of ribose at the cleft between two domains causes a conformational change corresponding to a closure of two domains around the ligand. The RBP has been crystallized in the open and closed conformations.
With the complex trajectory as a control, our goal was to study the conformation changes induced by the detachment of the ligand, and the results have been revealed from two computational tools, MD simulations and elastic network models.
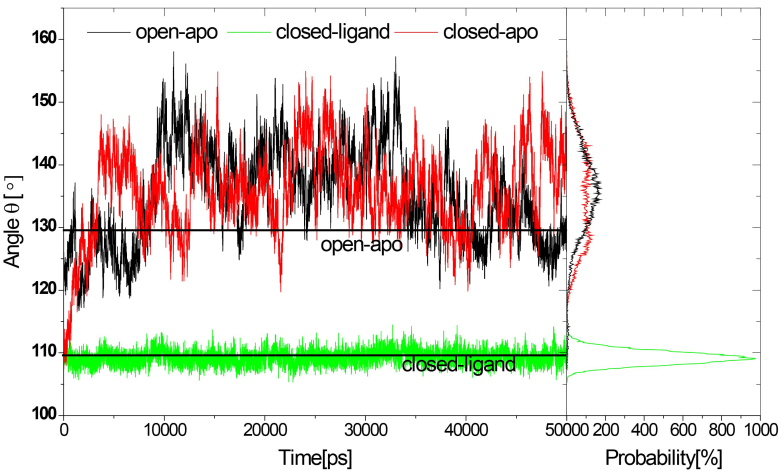
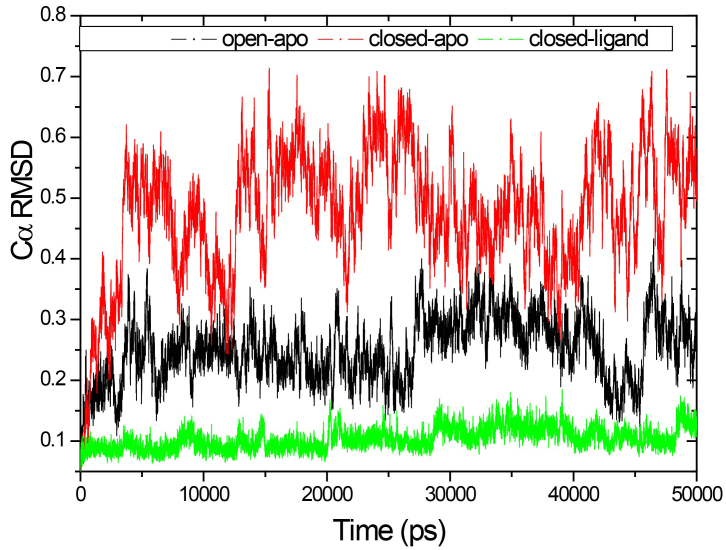
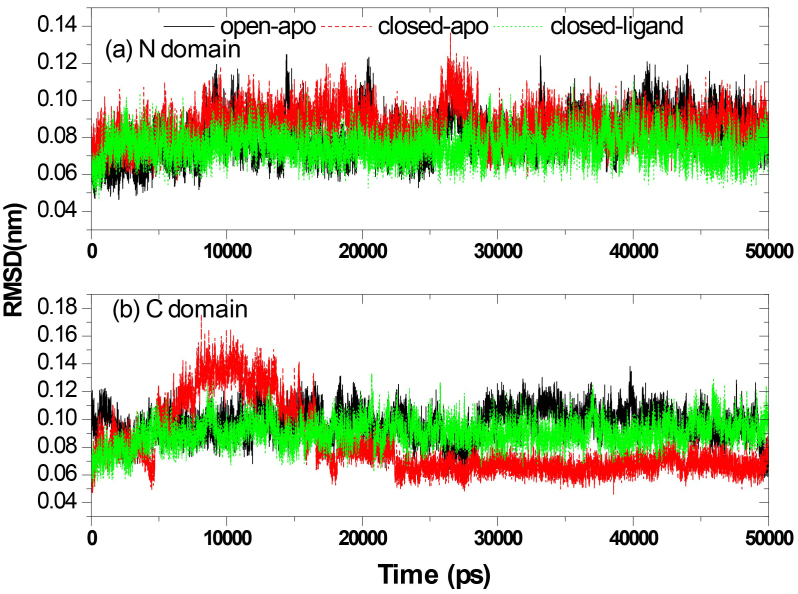
Molecular dynamics (MD) simulations were performed to study the conformation changes of RBP starting from the open-apo, closed-holo and closed-apo conformations.
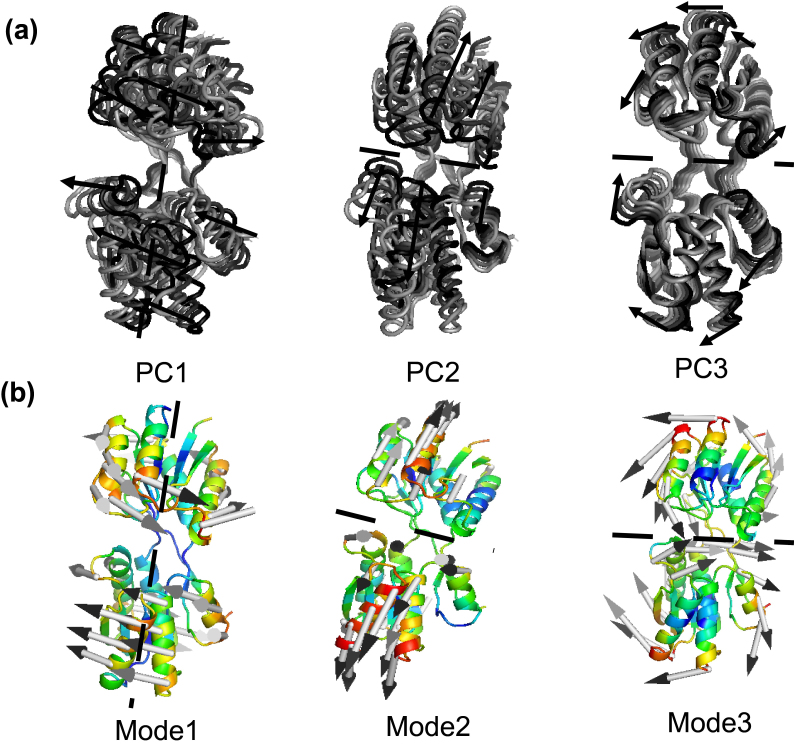
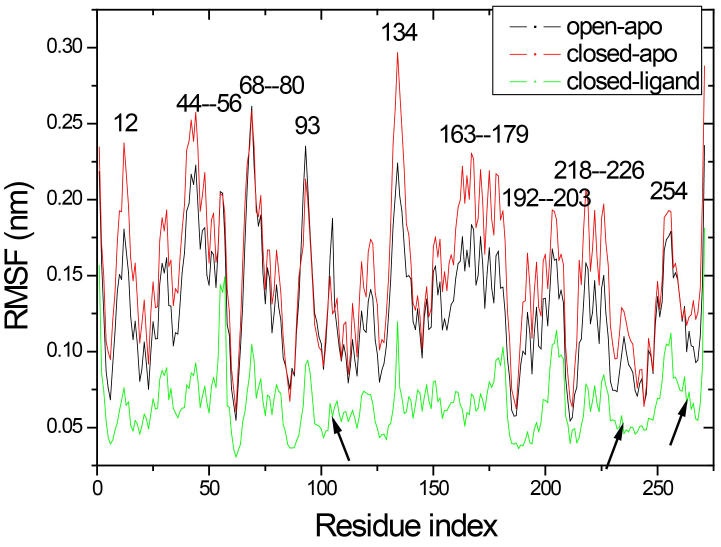
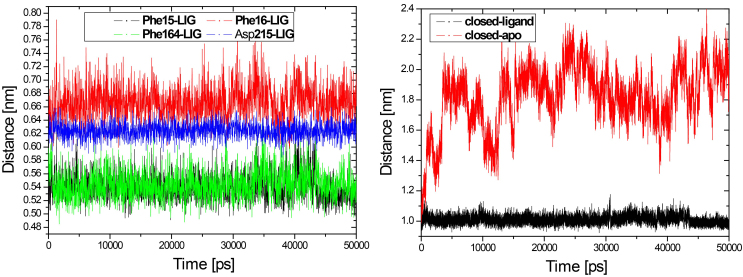
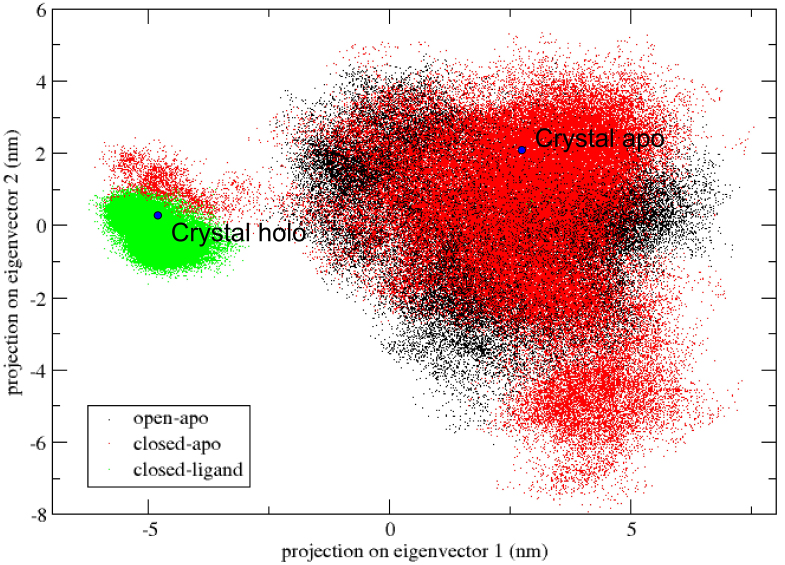
The evolution of the domain opening angle θ clearly indicates large structural changes. The simulations indicate that the closed states in the absence of ribose are inclined to transition to the open states and that ribose-free RBP exists in a wide range of conformations. The first three dominant principal motions derived from the closed-apo trajectories, consisting of rotating, bending and twisting motions, account for the major rearrangement of the domains from the closed to the open conformation.
The motions showed a strong one-to-one correspondence with the slowest modes from our previous study of RBP with the anisotropic network model (ANM). The results obtained for RBP contribute to the generalization of robustness for protein domain motion studies using either the ANM or PCA for trajectories obtained from MD.
大肠杆菌的核糖结合蛋白(RBP)是周质结合蛋白的代表性结构之一。在两个结构域之间的裂缝处结合核糖会导致构象变化,对应于两个结构域围绕配体的闭合。RBP 已经在开放和闭合构象中结晶。
以复杂轨迹为对照,我们的目标是研究配体脱离引起的构象变化,研究结果来自两种计算工具,分子动力学(MD)模拟和弹性网络模型。
进行分子动力学(MD)模拟,以研究从开放 apo、闭合 holo 和闭合 apo 构象开始的 RBP 构象变化。
域开口角度θ的演变清楚地表明了较大的结构变化。模拟表明,在没有核糖的情况下,闭合状态倾向于向开放状态转变,并且无核糖的 RBP 存在于广泛的构象范围内。从闭合 apo 轨迹得出的前三个主要主运动,由旋转、弯曲和扭曲运动组成,解释了结构域从闭合到开放构象的主要重排。
这些运动与我们之前使用各向异性网络模型(ANM)研究 RBP 时的最慢模式表现出很强的一一对应关系。所得结果有助于使用 MD 获得的轨迹的 ANM 或 PCA 来推广蛋白质结构域运动研究的稳健性。