Centre for Genomics Research, Discovery Sciences, BioPharmaceuticals R&D, AstraZeneca, Cambridge, UK.
National Center for Biotechnology Information, National Library of Medicine, Bethesda, MD, USA.
Nat Commun. 2021 Mar 8;12(1):1504. doi: 10.1038/s41467-021-21790-4.
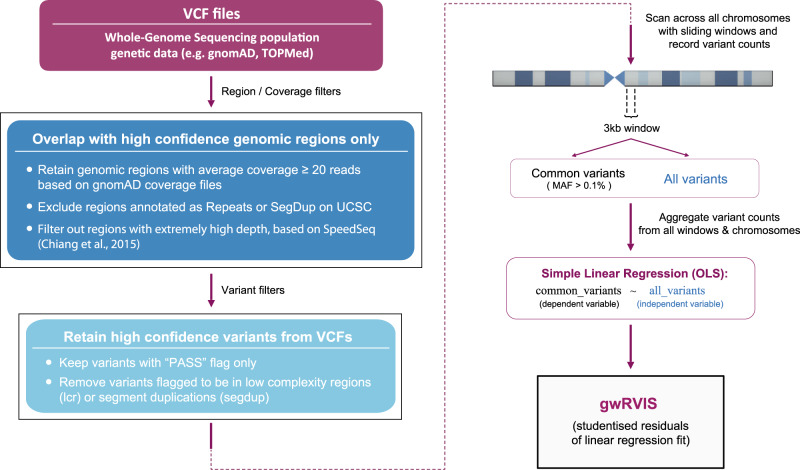
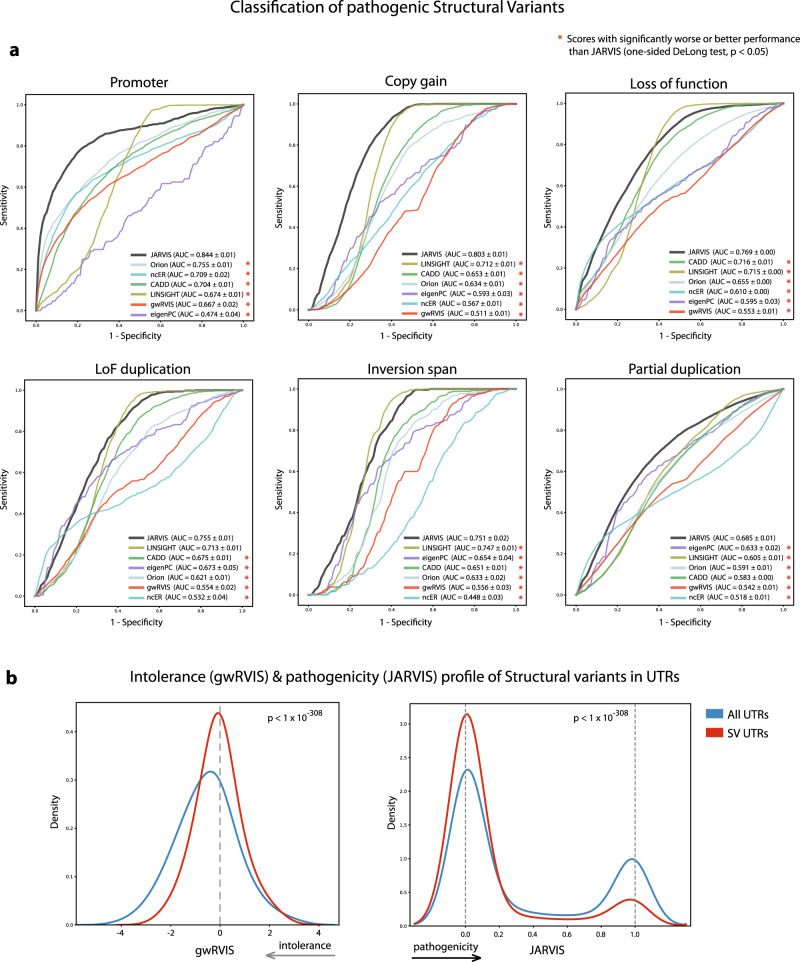
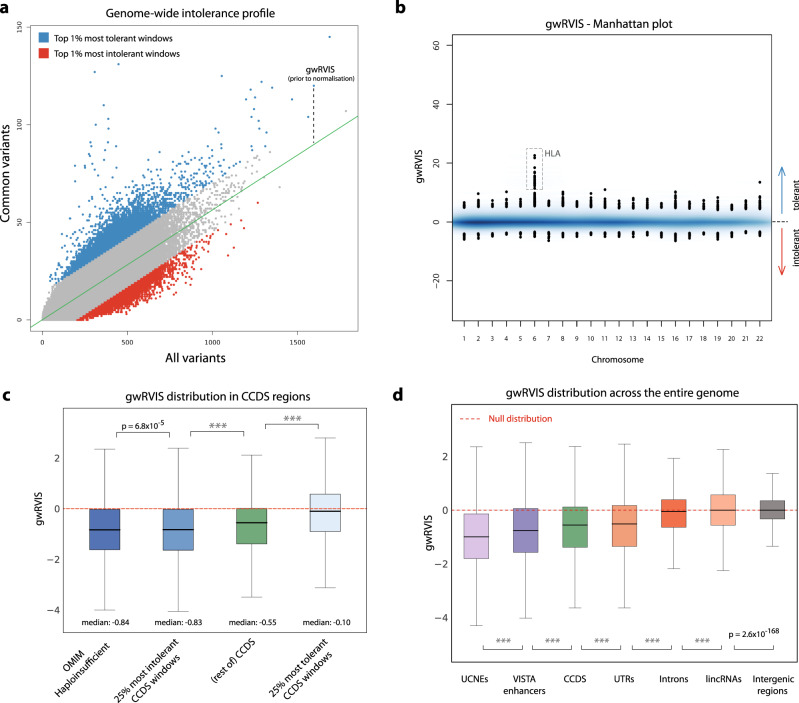
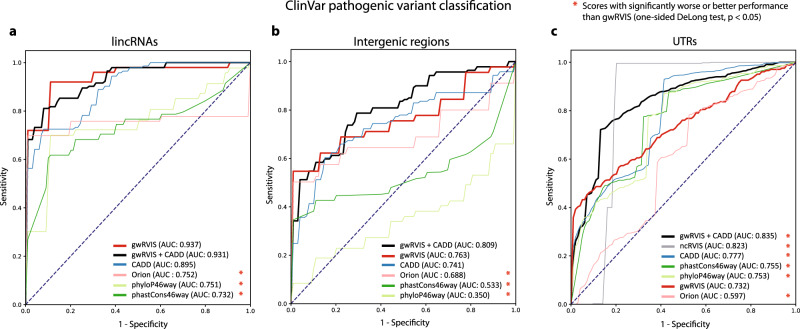
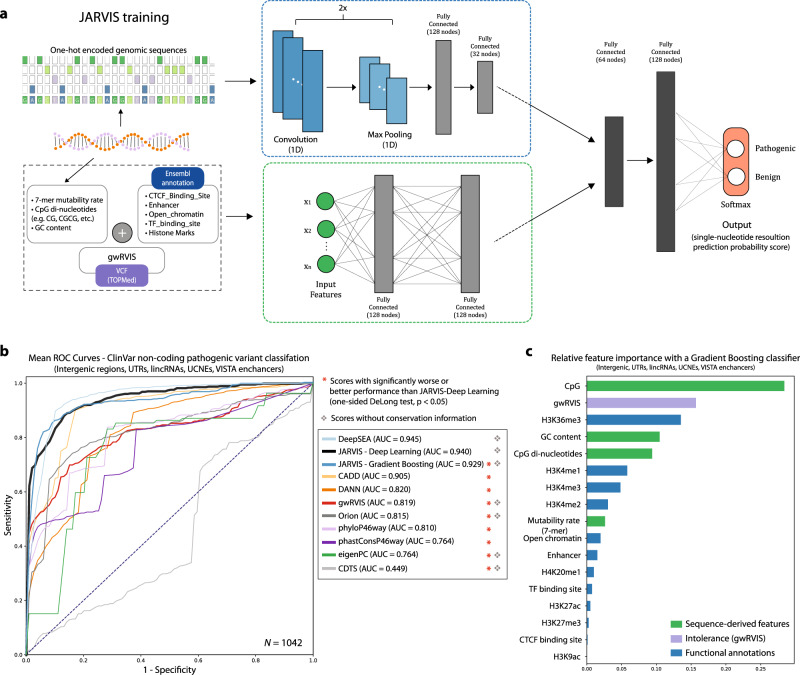
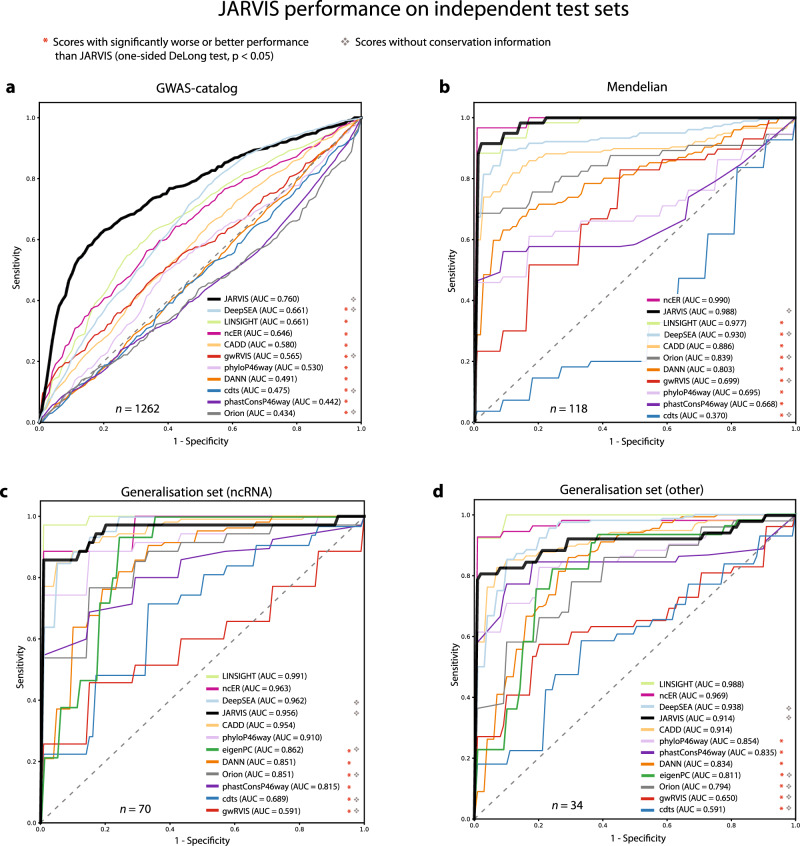
Elucidating functionality in non-coding regions is a key challenge in human genomics. It has been shown that intolerance to variation of coding and proximal non-coding sequence is a strong predictor of human disease relevance. Here, we integrate intolerance to variation, functional genomic annotations and primary genomic sequence to build JARVIS: a comprehensive deep learning model to prioritize non-coding regions, outperforming other human lineage-specific scores. Despite being agnostic to evolutionary conservation, JARVIS performs comparably or outperforms conservation-based scores in classifying pathogenic single-nucleotide and structural variants. In constructing JARVIS, we introduce the genome-wide residual variation intolerance score (gwRVIS), applying a sliding-window approach to whole genome sequencing data from 62,784 individuals. gwRVIS distinguishes Mendelian disease genes from more tolerant CCDS regions and highlights ultra-conserved non-coding elements as the most intolerant regions in the human genome. Both JARVIS and gwRVIS capture previously inaccessible human-lineage constraint information and will enhance our understanding of the non-coding genome.
阐明非编码区域的功能是人类基因组学的一个关键挑战。已经表明,对编码和近端非编码序列变异的不宽容是人类疾病相关性的一个强有力的预测因子。在这里,我们整合了变异的不宽容、功能基因组注释和主要基因组序列,构建了 JARVIS:一个全面的深度学习模型,用于对非编码区域进行优先级排序,优于其他人类谱系特异性评分。尽管 JARVIS 对进化保守性是不可知的,但在对致病性单核苷酸和结构变异进行分类时,JARVIS 的表现与基于保守性的评分相当或优于基于保守性的评分。在构建 JARVIS 时,我们引入了全基因组剩余变异不宽容评分(gwRVIS),该评分应用于来自 62784 个人的全基因组测序数据的滑动窗口方法。gwRVIS 将孟德尔疾病基因与更宽容的 CCDS 区域区分开来,并突出了超保守的非编码元件是人类基因组中最不宽容的区域。JARVIS 和 gwRVIS 都捕捉到了以前无法获得的人类谱系约束信息,将增强我们对非编码基因组的理解。