Internal Medicine D, Department of Nephrology, Hypertension and Rheumatology, and Interdisciplinary Fabry Center Münster (IFAZ), University Hospital Münster, Albert-Schweitzer-Campus 1, 48149, Münster, Germany.
Drugs. 2021 Apr;81(6):635-645. doi: 10.1007/s40265-021-01486-1. Epub 2021 Mar 15.
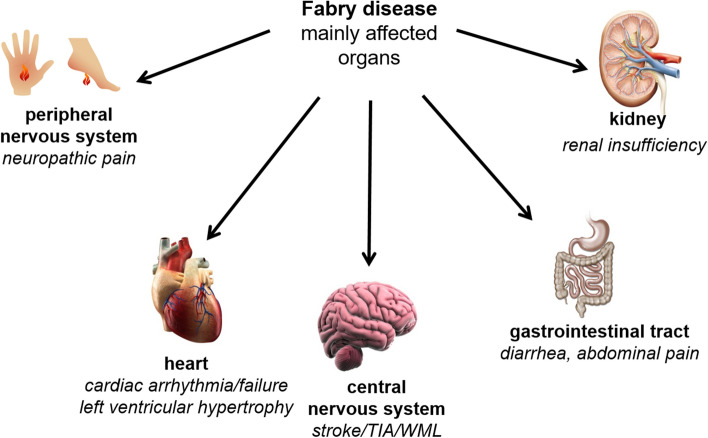
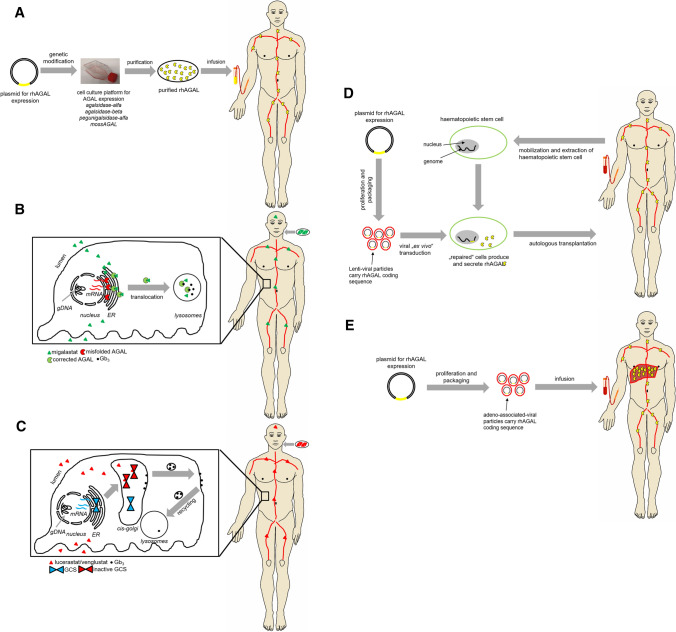
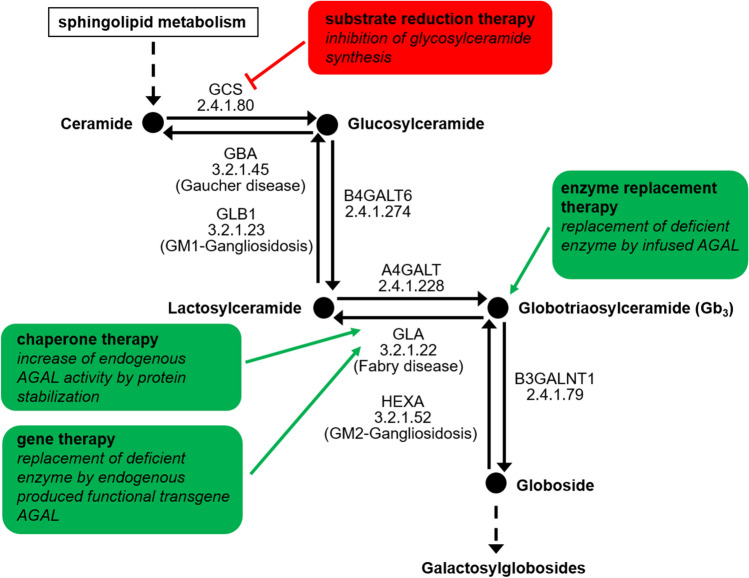
Fabry disease (FD) is a rare X-linked lysosomal storage disease based on a deficiency of α-galactosidase A (AGAL) caused by mutations in the α-galactosidase A gene (GLA). The lysosomal accumulation of glycosphingolipids, especially globotriaosylceramide (Gb) and globotriaosylsphingosine (lyso-Gb, deacylated form), leads to a multisystemic disease with progressive renal failure, cardiomyopathy with potentially malignant cardiac arrhythmias, and strokes, which considerably limits the life expectancy of affected patients. Diagnostic confirmation in male patients is based on the detection of AGAL deficiency in blood leukocytes, whereas in women, due to the potentially high residual enzymatic activity, molecular genetic detection of a causal mutation is required. Current treatment options for FD include recombinant enzyme replacement therapy (ERT) with intravenous agalsidase-alfa (0.2 mg/kg body weight) or agalsidase-beta (1 mg/kg body weight) every 2 weeks and oral chaperone therapy with migalastat (123 mg every other day), which selectively and reversibly binds to the active site of AGAL, thereby correcting the misfolding of the enzyme and allowing it to traffic to the lysosome. These therapies enable cellular Gb clearance and improve the burden of disease. However, in about 40% of all ERT-treated men, ERT can lead to infusion-associated reactions and the formation of neutralizing antidrug antibodies, which reduces the efficacy of therapy. In chaperone therapy, there are carriers of amenable mutations that show limited clinical success. This article provides a brief overview of the clinical picture in FD patients, diagnostic confirmation, and interdisciplinary clinical management of FD. The focus is on current and future therapeutic options.
法布里病(FD)是一种罕见的 X 连锁溶酶体贮积病,其病因是α-半乳糖苷酶 A(AGAL)基因(GLA)突变导致 α-半乳糖苷酶 A 缺乏。溶酶体中糖鞘脂,尤其是神经节苷脂(Gb)和神经节苷脂溶血(去酰化形式,lyso-Gb)的积累,导致多系统疾病,包括进行性肾衰竭、可能伴有恶性心律失常的心肌病和中风,这极大地限制了受影响患者的预期寿命。男性患者的诊断确认基于血液白细胞中 AGAL 缺乏的检测,而在女性中,由于潜在的高残留酶活性,需要进行分子遗传学检测以发现因果突变。FD 的当前治疗选择包括重组酶替代疗法(ERT),即每 2 周静脉给予阿加糖酶-α(0.2mg/kg 体重)或阿加糖酶-β(1mg/kg 体重),以及口服伴侣分子米加司他治疗(每隔一天给予 123mg),它选择性且可逆地与 AGAL 的活性部位结合,从而纠正酶的错误折叠并使其能够运输到溶酶体。这些疗法可使细胞内 Gb 清除,并改善疾病负担。然而,在接受 ERT 治疗的所有男性中,约有 40%会出现输注相关反应和中和性抗药物抗体的形成,从而降低治疗效果。在伴侣分子治疗中,有可治疗突变的载体,但临床疗效有限。本文简要概述了 FD 患者的临床表现、诊断确认以及 FD 的跨学科临床管理,重点是当前和未来的治疗选择。