Discovery Analytical, Medicinal Science and Technology, GlaxoSmithKline, Collegeville, Pennsylvania, USA.
Oncology R&D, GlaxoSmithKline, Collegeville, Pennsylvania, USA.
Mol Cell Proteomics. 2021;20:100067. doi: 10.1016/j.mcpro.2021.100067. Epub 2021 Mar 26.
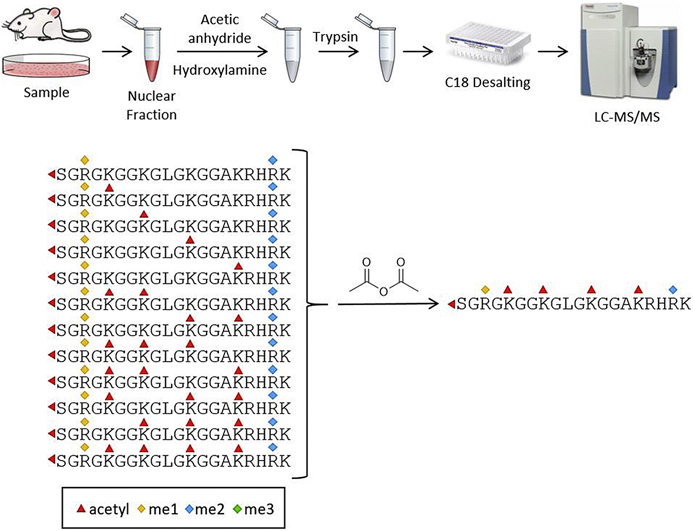
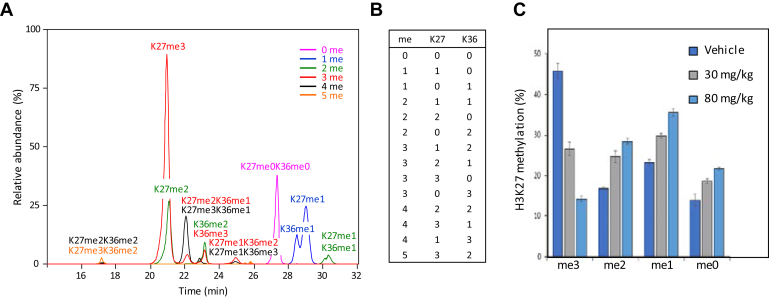
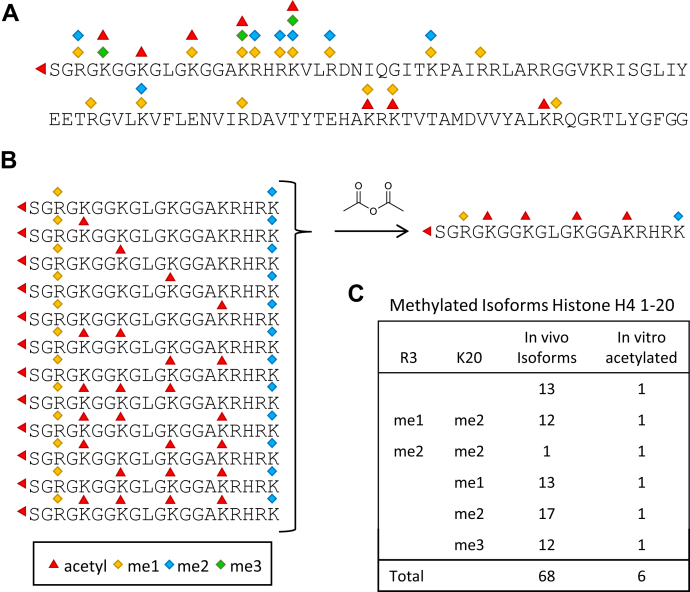
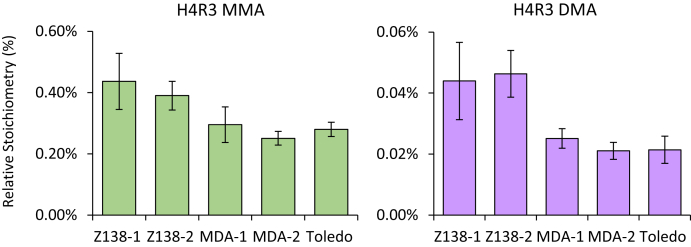
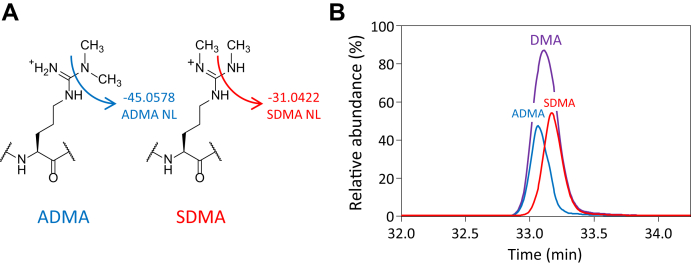
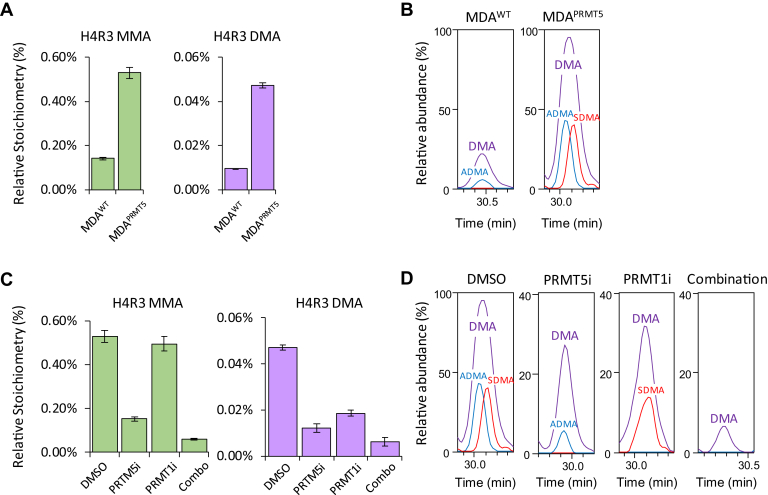
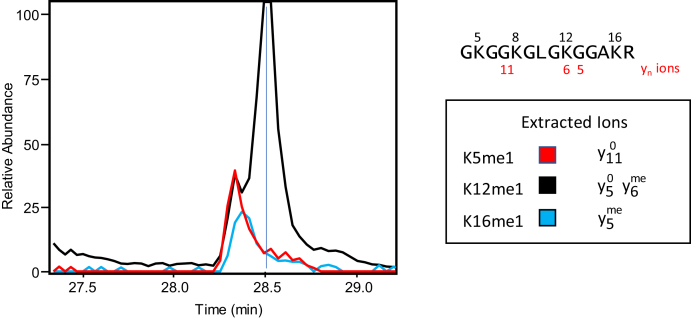
Histones are highly posttranslationally modified proteins that regulate gene expression by modulating chromatin structure and function. Acetylation and methylation are the most abundant histone modifications, with methylation occurring on lysine (mono-, di-, and trimethylation) and arginine (mono- and dimethylation) predominately on histones H3 and H4. In addition, arginine dimethylation can occur either symmetrically (SDMA) or asymmetrically (ADMA) conferring different biological functions. Despite the importance of histone methylation on gene regulation, characterization and quantitation of this modification have proven to be quite challenging. Great advances have been made in the analysis of histone modification using both bottom-up and top-down mass spectrometry (MS). However, MS-based analysis of histone posttranslational modifications (PTMs) is still problematic, due both to the basic nature of the histone N-terminal tails and to the combinatorial complexity of the histone PTMs. In this report, we describe a simplified MS-based platform for histone methylation analysis. The strategy uses chemical acetylation with d-acetic anhydride to collapse all the differently acetylated histone forms into one form, greatly reducing the complexity of the peptide mixture and improving sensitivity for the detection of methylation via summation of all the differently acetylated forms. We have used this strategy for the robust identification and relative quantitation of H4R3 methylation, for which stoichiometry and symmetry status were determined, providing an antibody-independent evidence that H4R3 is a substrate for both Type I and Type II PRMTs. Additionally, this approach permitted the robust detection of H4K5 monomethylation, a very low stoichiometry methylation event (0.02% methylation). In an independent example, we developed an in vitro assay to profile H3K27 methylation and applied it to an EZH2 mutant xenograft model following small-molecule inhibition of the EZH2 methyltransferase. These specific examples highlight the utility of this simplified MS-based approach to quantify histone methylation profiles.
组蛋白是高度翻译后修饰的蛋白质,通过调节染色质结构和功能来调节基因表达。乙酰化和甲基化是最丰富的组蛋白修饰,甲基化主要发生在赖氨酸(单、二和三甲基化)和精氨酸(单和二甲基化)上,主要发生在组蛋白 H3 和 H4 上。此外,精氨酸二甲基化可以对称(SDMA)或不对称(ADMA)发生,赋予不同的生物学功能。尽管组蛋白甲基化对基因调控很重要,但这种修饰的特征描述和定量已被证明极具挑战性。在使用自上而下和自下而上的质谱(MS)分析组蛋白修饰方面已经取得了重大进展。然而,基于 MS 的组蛋白翻译后修饰(PTM)分析仍然存在问题,这既是由于组蛋白 N 端尾部的碱性性质,也是由于组蛋白 PTM 的组合复杂性。在本报告中,我们描述了一种简化的基于 MS 的组蛋白甲基化分析平台。该策略使用 d-乙酸酐进行化学乙酰化,将所有不同乙酰化的组蛋白形式缩合成一种形式,大大降低了肽混合物的复杂性,并通过对所有不同乙酰化形式的总和提高了检测甲基化的灵敏度。我们已经使用该策略对 H4R3 甲基化进行了稳健的鉴定和相对定量,确定了其化学计量和对称状态,提供了 H4R3 是 I 型和 II 型 PRMTs 底物的抗体独立证据。此外,该方法还能够稳健地检测到 H4K5 单甲基化,这是一种非常低的化学计量甲基化事件(0.02%甲基化)。在一个独立的例子中,我们开发了一种体外测定法来分析 H3K27 甲基化,并将其应用于 EZH2 甲基转移酶的小分子抑制后 EZH2 突变体异种移植模型。这些具体示例突出了这种简化的基于 MS 的方法在定量组蛋白甲基化谱中的实用性。