Department of Pathology, Qingdao Municipal Hospital (Group), Qingdao, China.
Guangdong Provincial Key Laboratory of Shock and Microcirculation, School of Basic Medical Sciences, Southern Medical University, Guangzhou, China.
Front Immunol. 2021 Mar 12;12:625627. doi: 10.3389/fimmu.2021.625627. eCollection 2021.
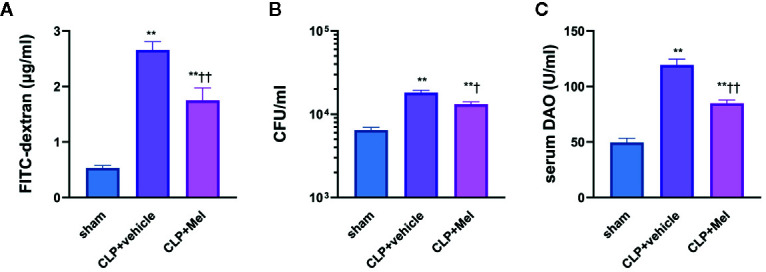
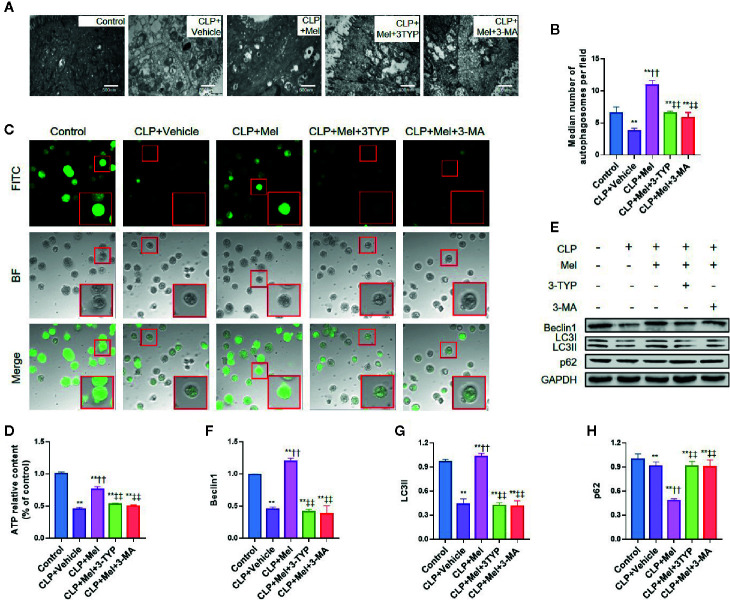
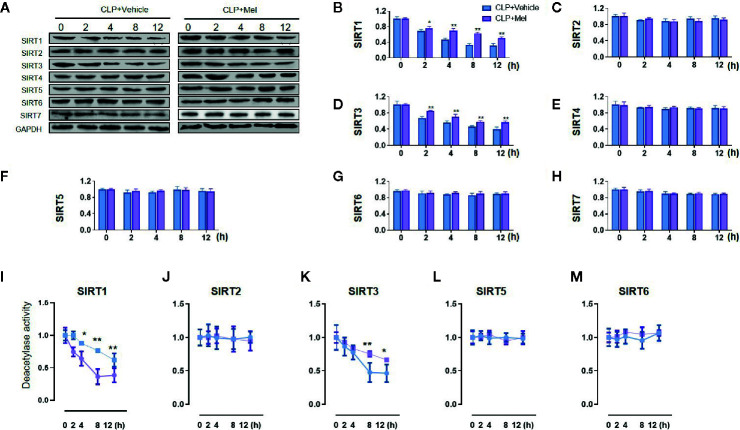
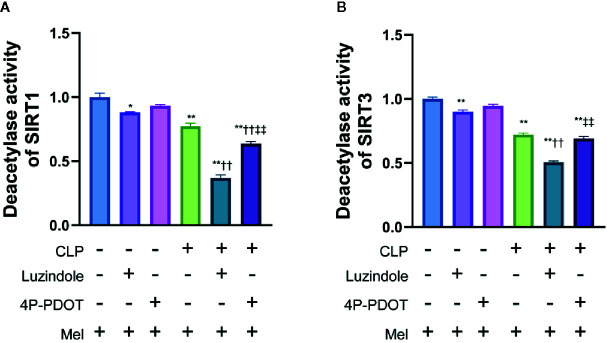
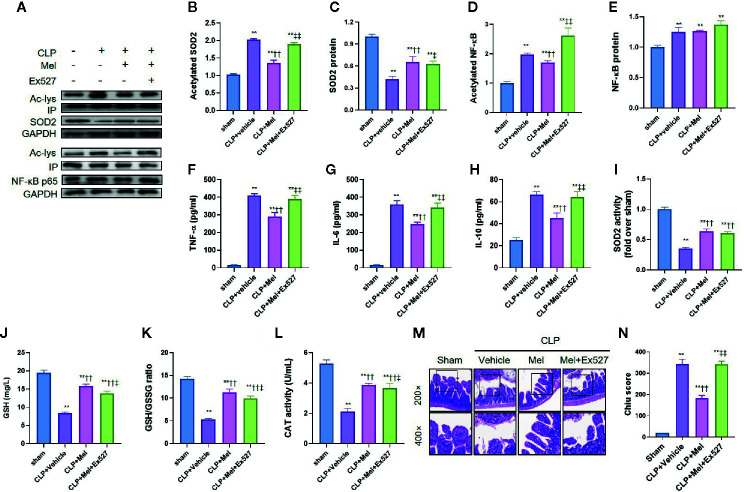
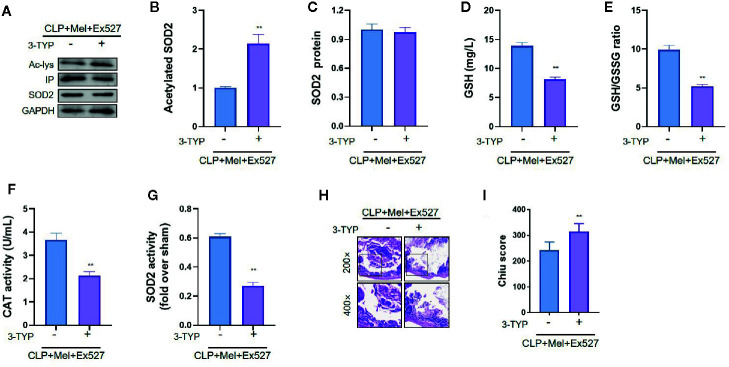
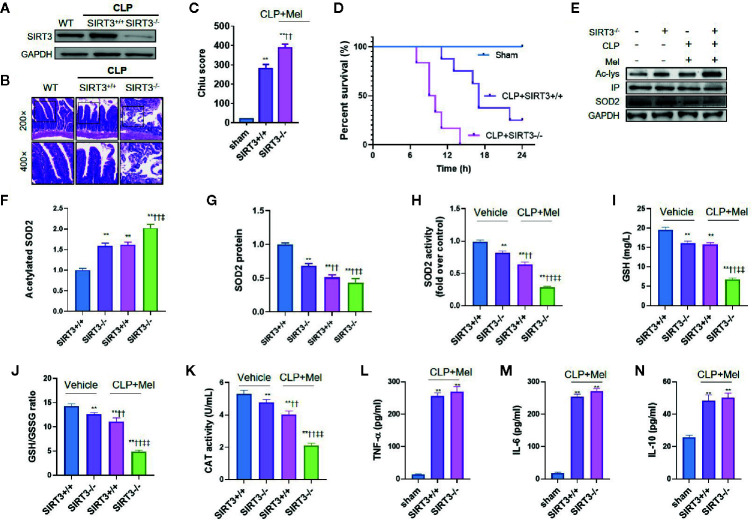
Melatonin reportedly alleviates sepsis-induced multi-organ injury by inducing autophagy and activating class III deacetylase Sirtuin family members (SIRT1-7). However, whether melatonin attenuates small-intestine injury along with the precise underlying mechanism remain to be elucidated. To investigate this, we employed cecal ligation and puncture (CLP)- or endotoxemia-induced sepsis mouse models and confirmed that melatonin treatment significantly prolonged the survival time of mice and ameliorated multiple-organ injury (lung/liver/kidney/small intestine) following sepsis. Melatonin partially protected the intestinal barrier function and restored SIRT1 and SIRT3 activity/protein expression in the small intestine. Mechanistically, melatonin treatment enhanced NF-κB deacetylation and subsequently reduced the inflammatory response and decreased the TNF-α, IL-6, and IL-10 serum levels; these effects were abolished by SIRT1 inhibition with the selective blocker, Ex527. Correspondingly, melatonin treatment triggered SOD2 deacetylation and increased SOD2 activity and subsequently reduced oxidative stress; this amelioration of oxidative stress by melatonin was blocked by the SIRT3-selective inhibitor, 3-TYP, and was independent of SIRT1. We confirmed this mechanistic effect in a CLP-induced sepsis model of intestinal SIRT3 conditional-knockout mice, and found that melatonin preserved mitochondrial function and induced autophagy of small-intestine epithelial cells; these effects were dependent on SIRT3 activation. This study has shown, to the best of our knowledge, for the first time that melatonin alleviates sepsis-induced small-intestine injury, at least partially, by upregulating SIRT3-mediated oxidative-stress inhibition, mitochondrial-function protection, and autophagy induction.
据报道,褪黑素通过诱导自噬和激活 III 类去乙酰化酶 Sirtuin 家族成员(SIRT1-7)来减轻脓毒症引起的多器官损伤。然而,褪黑素是否能减轻小肠损伤以及确切的潜在机制仍有待阐明。为了研究这一点,我们使用盲肠结扎和穿孔(CLP)或内毒素血症诱导的脓毒症小鼠模型,并证实褪黑素治疗显著延长了脓毒症小鼠的生存时间,并改善了多器官损伤(肺/肝/肾/小肠)。褪黑素部分保护了肠道屏障功能,并恢复了小肠中的 SIRT1 和 SIRT3 活性/蛋白表达。在机制上,褪黑素处理增强了 NF-κB 的去乙酰化,随后减少了炎症反应,并降低了 TNF-α、IL-6 和 IL-10 的血清水平;这些作用被 SIRT1 抑制剂 Ex527 选择性阻断。相应地,褪黑素处理触发了 SOD2 的去乙酰化,增加了 SOD2 的活性,随后减少了氧化应激;褪黑素对氧化应激的这种改善作用被 SIRT3 选择性抑制剂 3-TYP 阻断,并且独立于 SIRT1。我们在 CLP 诱导的脓毒症肠道 SIRT3 条件性敲除小鼠模型中证实了这种机制作用,并发现褪黑素维持了线粒体功能并诱导了小肠上皮细胞的自噬;这些作用依赖于 SIRT3 的激活。这项研究首次表明,褪黑素通过上调 SIRT3 介导的氧化应激抑制、线粒体功能保护和自噬诱导,至少部分缓解了脓毒症引起的小肠损伤。