Department of Otolaryngology, National Taiwan University Hospital, Taipei 100, Taiwan.
Program in Speech and Hearing Bioscience and Technology, Harvard Medical School, Boston, MA 02115, USA.
Int J Mol Sci. 2021 Mar 10;22(6):2789. doi: 10.3390/ijms22062789.
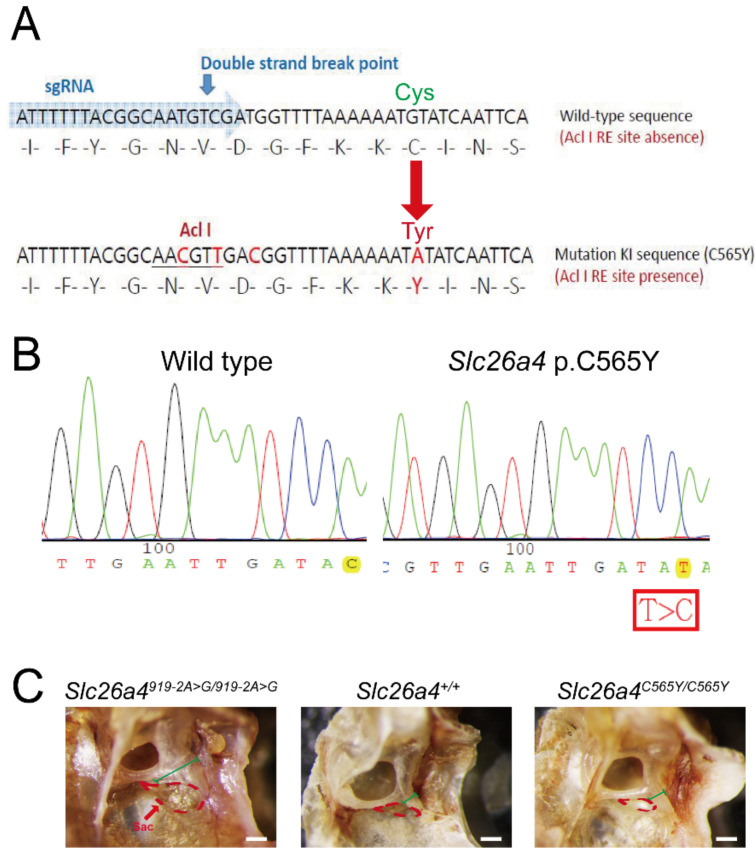
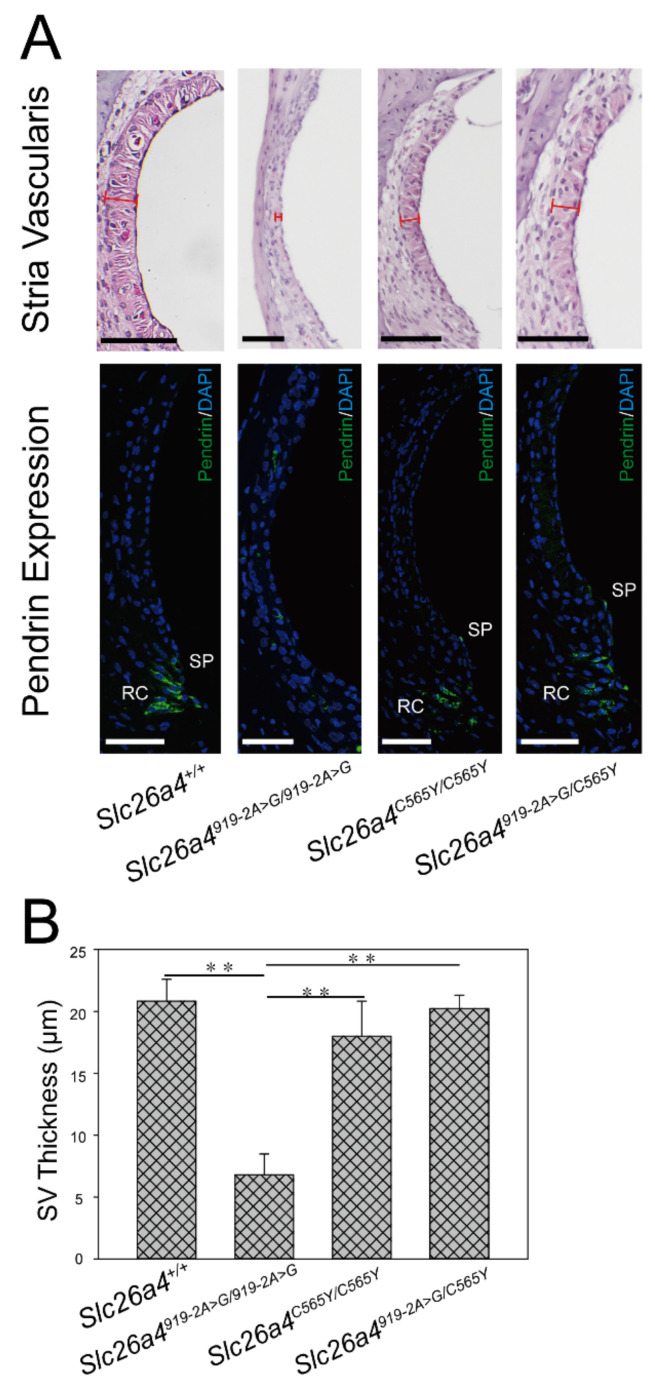
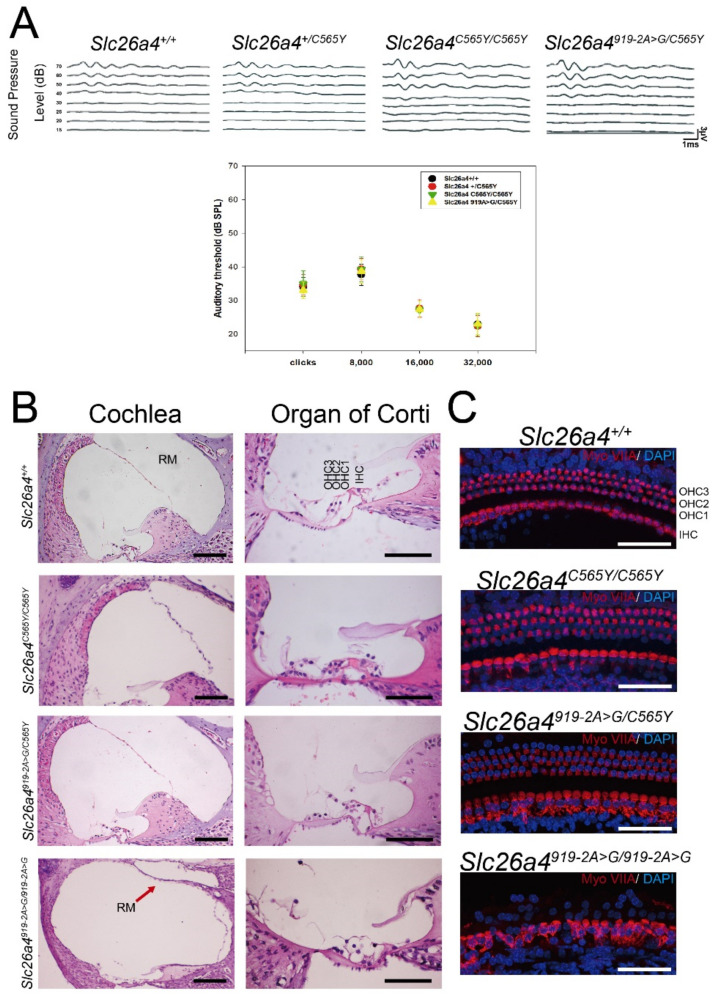
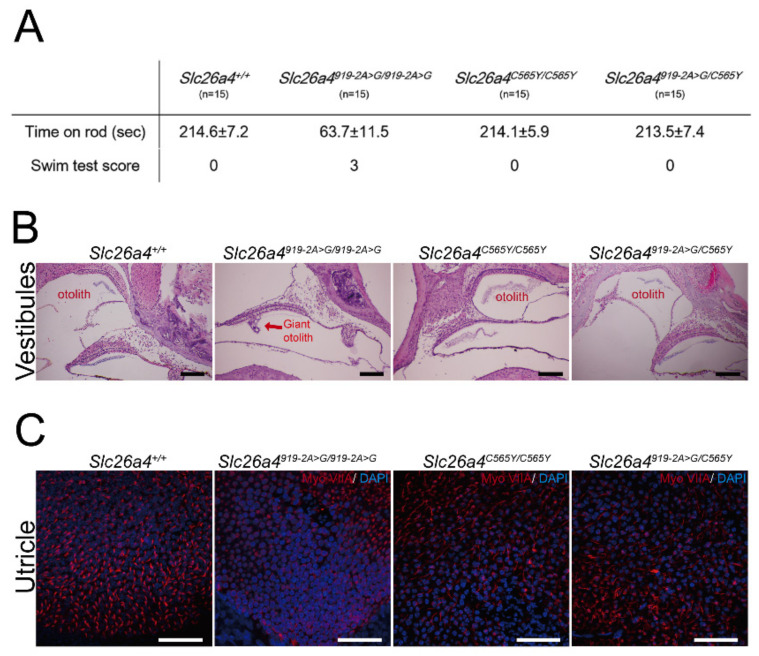
Recessive variants of the gene are globally a common cause of hearing impairment. In the past, cell lines and transgenic mice were widely used to investigate the pathogenicity associated with variants. However, discrepancies in pathogenicity between humans and cell lines or transgenic mice were documented for some variants. For instance, the p.C565Y variant, which was reported to be pathogenic in humans, did not exhibit functional pathogenic consequences in cell lines. To address the pathogenicity of p.C565Y, we used a genotype-based approach in which we generated knock-in mice that were heterozygous (), homozygous (), and compound heterozygous () for this variant. Subsequent phenotypic characterization revealed that mice with these genotypes demonstrated normal auditory and vestibular functions, and normal inner-ear morphology and pendrin expression. These findings indicate that the p.C565Y variant is nonpathogenic for mice, and that a single p.C565Y allele is sufficient to maintain normal inner-ear physiology in mice. Our results highlight the differences in pathogenicity associated with certain variants between transgenic mice and humans, which should be considered when interpreting the results of animal studies for -related deafness.
基因的隐性变异是全球听力障碍的常见原因。过去,细胞系和转基因小鼠被广泛用于研究与变体相关的致病性。然而,一些变体的人类与细胞系或转基因小鼠之间的致病性存在差异。例如,据报道在人类中致病性的 p.C565Y 变体在细胞系中没有表现出功能致病性后果。为了解决 p.C565Y 的致病性问题,我们使用了基于基因型的方法,在该方法中,我们生成了杂合子 ()、纯合子 () 和复合杂合子 () 的敲入小鼠。随后的表型特征表明,具有这些基因型的小鼠表现出正常的听觉和前庭功能,以及正常的内耳形态和 pendrin 表达。这些发现表明,p.C565Y 变体对小鼠是非致病性的,并且单个 p.C565Y 等位基因足以维持小鼠内耳生理学的正常。我们的结果强调了某些变体在转基因小鼠和人类之间与致病性相关的差异,在解释与相关耳聋的动物研究结果时应考虑这些差异。