Edinburgh Medical School: Biomedical Sciences, University of Edinburgh, Edinburgh, EH8 9XD, UK.
Euan MacDonald Centre for Motor Neurone Disease Research, University of Edinburgh, Edinburgh, EH16 4SB, UK.
Cell Mol Life Sci. 2021 May;78(10):4785-4804. doi: 10.1007/s00018-021-03819-5. Epub 2021 Apr 5.
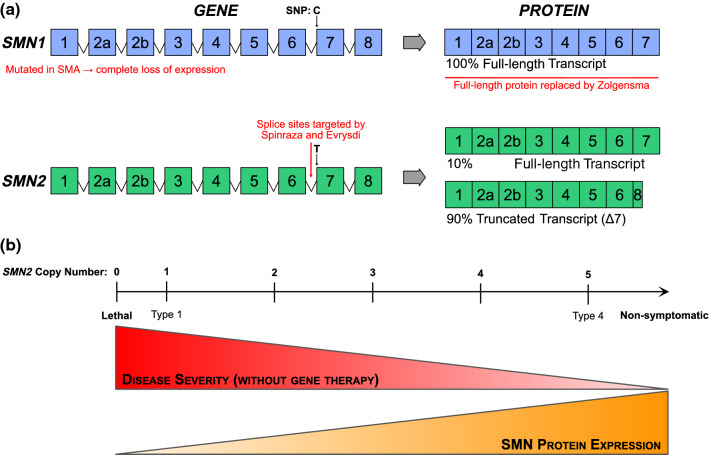
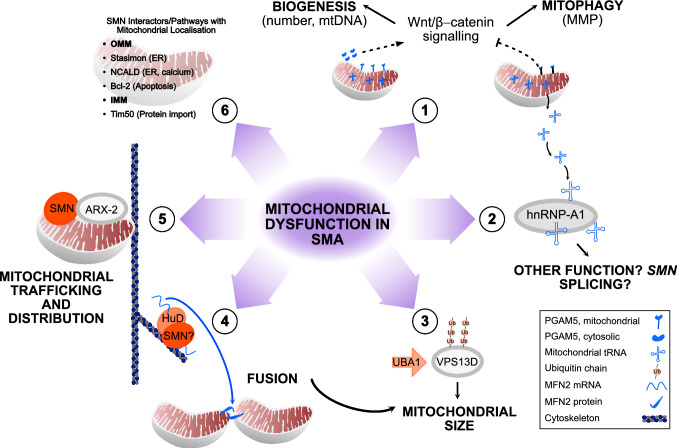
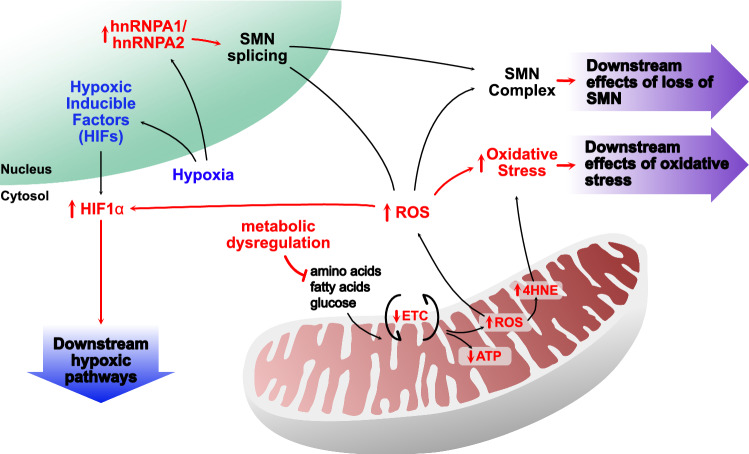
Spinal muscular atrophy (SMA) is an autosomal recessive motor neuron disease of variable clinical severity that is caused by mutations in the survival motor neuron 1 (SMN1) gene. Despite its name, SMN is a ubiquitous protein that functions within and outside the nervous system and has multiple cellular roles in transcription, translation, and proteostatic mechanisms. Encouragingly, several SMN-directed therapies have recently reached the clinic, albeit this has highlighted the increasing need to develop combinatorial therapies for SMA to achieve full clinical efficacy. As a subcellular site of dysfunction in SMA, mitochondria represents a relevant target for a combinatorial therapy. Accordingly, we will discuss our current understanding of mitochondrial dysfunction in SMA, highlighting mitochondrial-based pathways that offer further mechanistic insights into the involvement of mitochondria in SMA. This may ultimately facilitate translational development of targeted mitochondrial therapies for SMA. Due to clinical and mechanistic overlaps, such strategies may also benefit other motor neuron diseases and related neurodegenerative disorders.
脊髓性肌萎缩症(SMA)是一种由生存运动神经元 1(SMN1)基因突变引起的常染色体隐性运动神经元疾病,其临床严重程度不一。尽管它的名字是 SMA,但 SMN 是一种普遍存在的蛋白质,在神经系统内外都有功能,在转录、翻译和蛋白质稳态机制中具有多种细胞功能。令人鼓舞的是,最近有几种针对 SMA 的 SMN 靶向疗法已经进入临床阶段,尽管这凸显了开发 SMA 组合疗法以实现完全临床疗效的必要性日益增加。作为 SMA 中功能障碍的亚细胞部位,线粒体是组合治疗的一个相关靶点。因此,我们将讨论我们目前对 SMA 中线粒体功能障碍的理解,强调基于线粒体的途径,这些途径为线粒体参与 SMA 提供了进一步的机制见解。这可能最终有助于 SMA 的靶向线粒体治疗的转化发展。由于临床和机制上的重叠,这些策略也可能有益于其他运动神经元疾病和相关的神经退行性疾病。