Divyashri G, Krishna Murthy T P, Sundareshan Subramaniam, Kamath Pavan, Murahari Manikanta, Saraswathy G R, Sadanandan Bindu
Department of Biotechnology, M S Ramaiah Institute of Technology, Bangalore, Karnataka, India.
Pharmacological Modelling and Simulation Centre, M.S. Ramaiah University of Applied Sciences, Bangalore, Karnataka, India.
Bioimpacts. 2021;11(2):119-127. doi: 10.34172/bi.2021.19. Epub 2020 Mar 24.
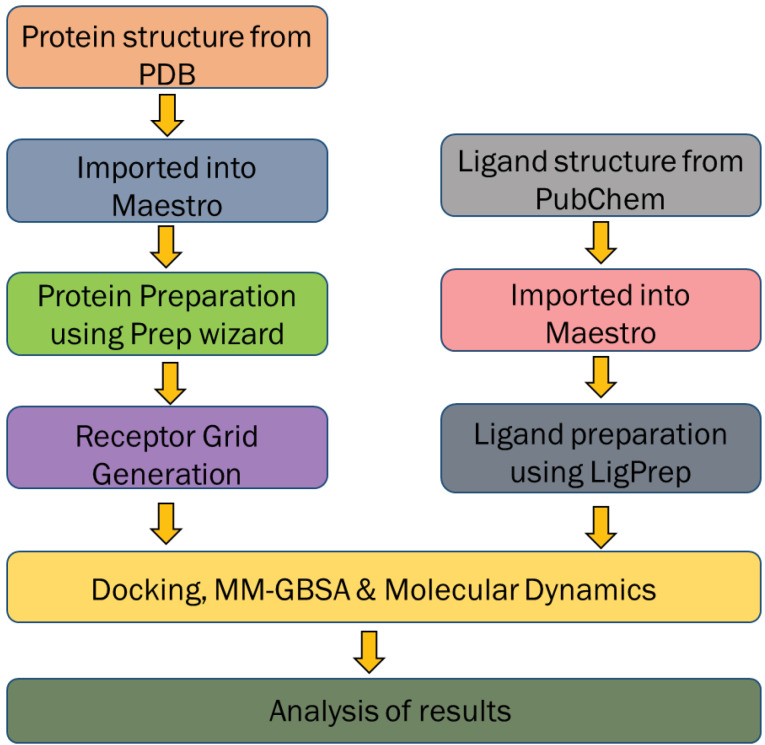
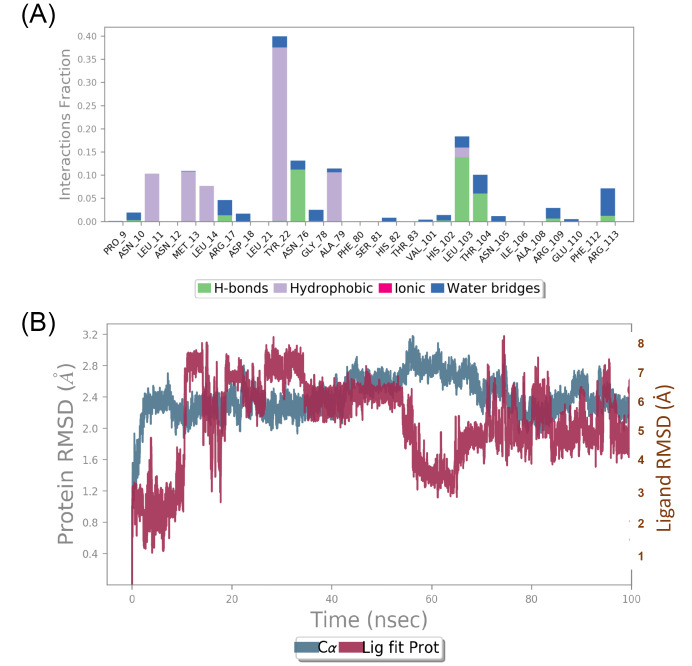
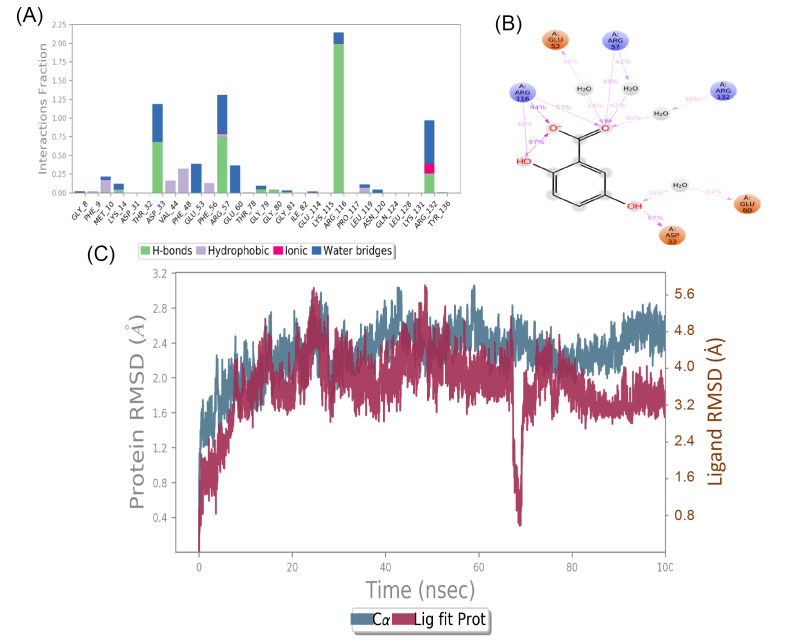
The present study attempts to identify potential targets of for novel inhibitors from therapeutic herb, mango ginger ( Roxb.). Crystal structure of all the selected drug targets obtained from Protein Data Bank (PDB) were subjected to molecular docking against a total of 130 compounds (found to have biological activity against ) were retrieved from public databases. Compounds with good binding affinity were selected for Prime MM-GBSA rescoring and molecular dynamics (MD) simulation. Final list of compounds were taken for ADMET predictions. Based on binding affinity denoted by glide score and ligand efficiency, mango ginger compounds were found selective to shikimate kinase and type II dehydroquinase through hydrogen bonding and salt bridge interactions. Stability of the interactions and free energy calculations by Prime MM-GBSA results confirmed the affinity of mango ginger compounds towards both shikimate kinase and type II dehydroquinase. From the above results, 15 compounds were calculated for ADMET parameters, Lipinski's rule of five, and the results were found promising without any limitations. MD simulations identified gentisic acid as hit compound for shikimate kinase of . Current study could identify the potential of mango ginger compounds against shikimate kinase and type II dehydroquinase targets for infections and are suitable for and evaluation.
本研究试图从药用植物芒果姜(Curcuma amada Roxb.)中鉴定新型抑制剂的潜在靶点。从蛋白质数据库(PDB)获得的所有选定药物靶点的晶体结构,针对从公共数据库中检索到的总共130种化合物(发现对[具体疾病]具有生物活性)进行分子对接。选择具有良好结合亲和力的化合物进行Prime MM - GBSA重新评分和分子动力学(MD)模拟。对最终的化合物列表进行ADMET预测。基于由Glide评分和配体效率表示的结合亲和力,发现芒果姜化合物通过氢键和盐桥相互作用对莽草酸激酶和II型脱氢奎尼酸酶具有选择性。通过Prime MM - GBSA结果进行的相互作用稳定性和自由能计算证实了芒果姜化合物对莽草酸激酶和II型脱氢奎尼酸酶的亲和力。根据上述结果,计算了15种化合物的ADMET参数、Lipinski五规则,结果显示前景良好且无任何限制。MD模拟确定龙胆酸为[具体疾病]莽草酸激酶的命中化合物。当前研究可以确定芒果姜化合物针对莽草酸激酶和II型脱氢奎尼酸酶靶点对[具体疾病]感染的潜在作用,并且适用于[具体方面1]和[具体方面2]评估。