He Mei-Nan, Zhao Shan-Chao, Li Ji-Min, Tong Lu-Lu, Fan Xin-Zhao, Xue Yao-Ming, Lin Xiao-Hong, Cao Ying
Department of Endocrinology and Metabolism, Nanfang Hospital, Southern Medical University, Guangzhou 510515, Guangdong Province, China.
Department of Urology, Nanfang Hospital, Southern Medical University, Guangzhou 510515, Guangdong Province, China.
World J Clin Cases. 2021 Apr 6;9(10):2259-2267. doi: 10.12998/wjcc.v9.i10.2259.
Co-morbidity of gene turner syndrome (TS) with positive gene and non-classical congenital adrenal hyperplasia (NCAH) is extremely rare and has never been reported to date.
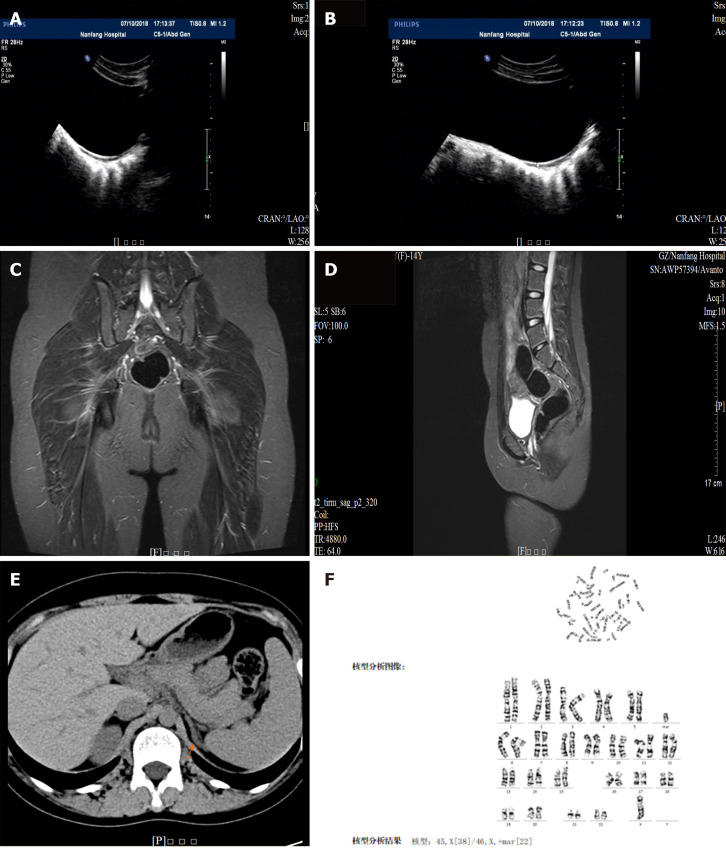
In this article, we present a 14-year-old girl who was referred to our hospital with short stature (weight of 43 kg and height of 143 cm, < -2 SD) with no secondary sexual characteristics (labia minora dysplasia). Laboratory tests indicated hypergonadotropic hypogonadism with significantly increased androstenedione and 17-hydroxyprogesterone (17-OHP) levels. This was accompanied by the thickening of the extremity of the left adrenal medial limb. The patient's karyotype was 45,X/46,X, +mar, and cytogenetic analysis using multiplex ligation-dependent probe amplification and high-throughput sequencing indicated that the gene was positive with compound heterozygous mutations in as the causative gene for congenital adrenal hyperplasia. The sites of the suspected candidate mutations were amplified and verified using Sanger sequencing. The patient was finally diagnosed as having positive TS with NCAH. The patient and her family initially refused medical treatment. At her most recent follow-up visit (age = 15 years old), the patient presented facial hair, height increase to 148 cm, and weight of 52 kg, while androstenedione and 17-OHP levels remained high. The patient was finally willing to take small doses of hydrocortisone (10 mg/d).
In conclusion, upon evaluation of the patient mentioned in the report, we feel that 17-OHP measurement and cytogenetic analysis are necessary for TS patients even in the absence of significant virilization signs. This will play a significant role in guiding diagnosis and treatment.
特纳综合征(TS)基因与阳性基因及非经典型先天性肾上腺皮质增生症(NCAH)的共病极为罕见,迄今为止尚未见报道。
在本文中,我们介绍了一名14岁女孩,因身材矮小(体重43千克,身高143厘米,<-2标准差)且无第二性征(小阴唇发育不良)转诊至我院。实验室检查显示高促性腺激素性性腺功能减退,雄烯二酮和17-羟孕酮(17-OHP)水平显著升高。同时伴有左肾上腺内侧肢末端增厚。患者的核型为45,X/46,X,+mar,采用多重连接依赖探针扩增和高通量测序进行细胞遗传学分析表明,该基因呈阳性,存在复合杂合突变,作为先天性肾上腺皮质增生症的致病基因。使用桑格测序法对疑似候选突变位点进行扩增和验证。该患者最终被诊断为阳性TS合并NCAH。患者及其家属最初拒绝治疗。在她最近一次随访(年龄15岁)时,患者出现面部毛发,身高增至148厘米,体重52千克,而雄烯二酮和17-OHP水平仍然很高。患者最终愿意服用小剂量氢化可的松(10毫克/天)。
总之,通过对报告中提及患者的评估,我们认为即使TS患者没有明显的男性化体征,进行17-OHP测量和细胞遗传学分析也是必要的。这将对指导诊断和治疗发挥重要作用。