Department of Chemical and Biomolecular Engineering, University of Delaware, Newark, Delaware, USA.
Ammon Pinizzotto Biopharmaceutical Innovation Center, Newark, Delaware, USA.
mSphere. 2021 Apr 21;6(2):e01336-20. doi: 10.1128/mSphere.01336-20.
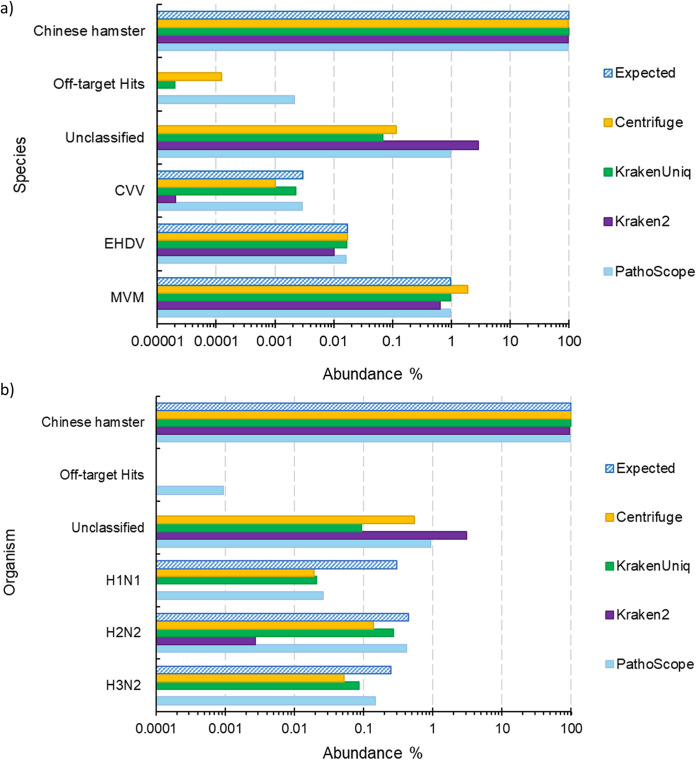
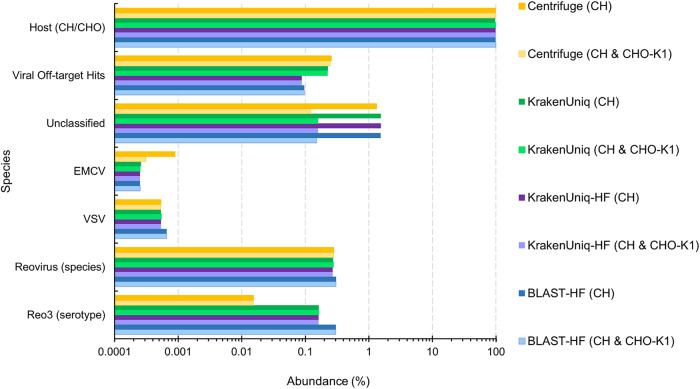
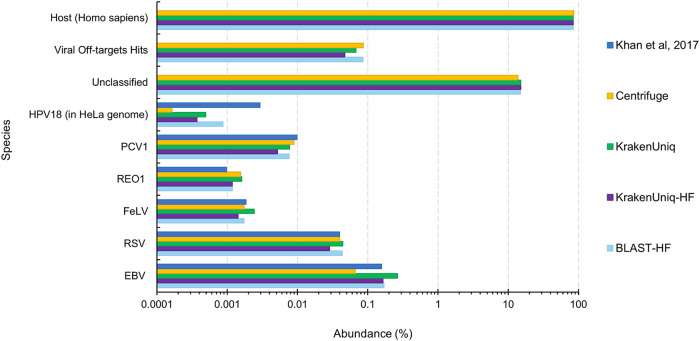
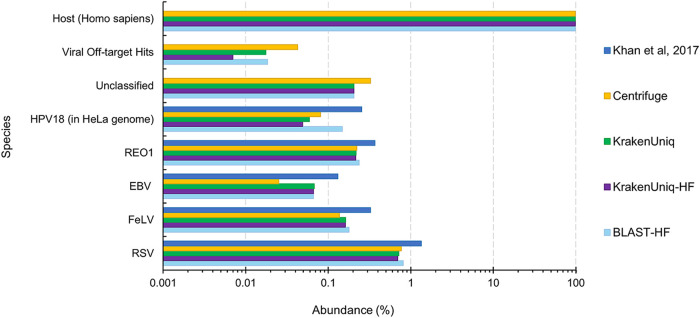
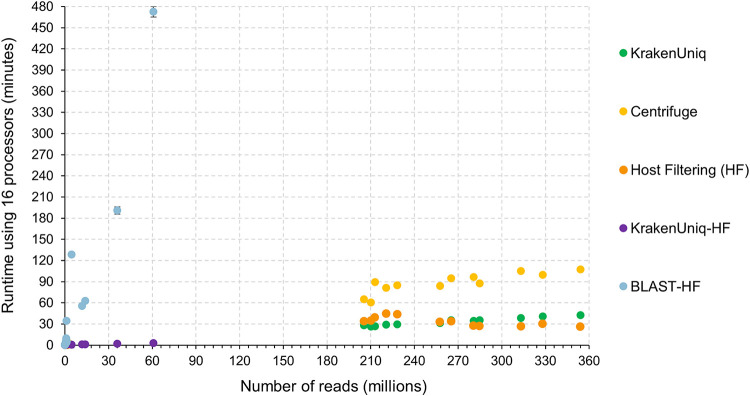
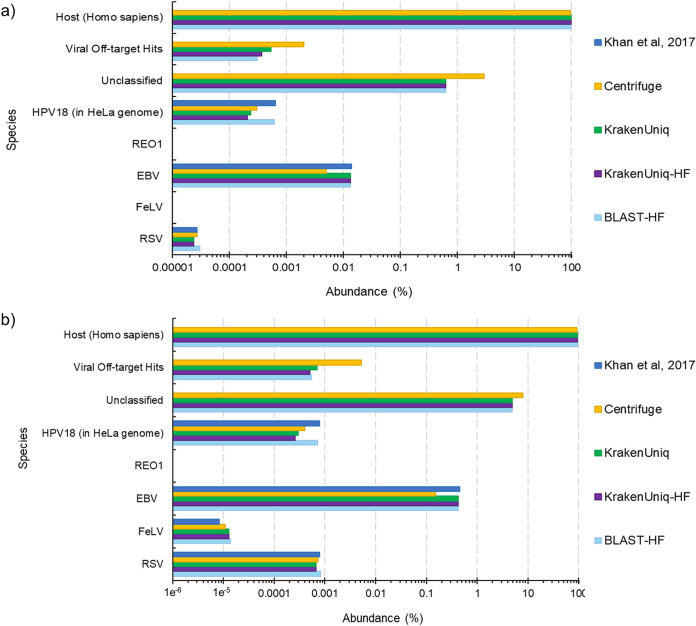
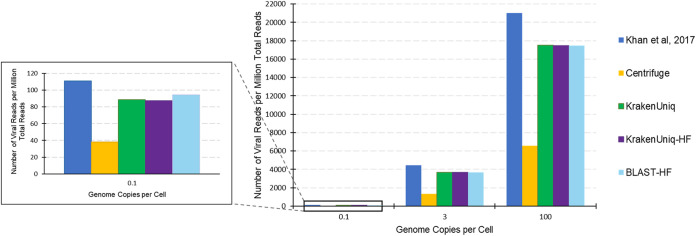
Adventitious agent detection during the production of vaccines and biotechnology-based medicines is of critical importance to ensure the final product is free from any possible viral contamination. Increasing the speed and accuracy of viral detection is beneficial as a means to accelerate development timelines and to ensure patient safety. Here, several rapid viral metagenomics approaches were tested on simulated next-generation sequencing (NGS) data sets and existing data sets from virus spike-in studies done in CHO-K1 and HeLa cell lines. It was observed that these rapid methods had comparable sensitivity to full-read alignment methods used for NGS viral detection for these data sets, but their specificity could be improved. A method that first filters host reads using KrakenUniq and then selects the virus classification tool based on the number of remaining reads is suggested as the preferred approach among those tested to detect nonlatent and nonendogenous viruses. Such an approach shows reasonable sensitivity and specificity for the data sets examined and requires less time and memory as full-read alignment methods. Next-generation sequencing (NGS) has been proposed as a complementary method to detect adventitious viruses in the production of biotherapeutics and vaccines to current and methods. Before NGS can be established in industry as a main viral detection technology, further investigation into the various aspects of bioinformatics analyses required to identify and classify viral NGS reads is needed. In this study, the ability of rapid metagenomics tools to detect viruses in biopharmaceutical relevant samples is tested and compared to recommend an efficient approach. The results showed that KrakenUniq can quickly and accurately filter host sequences and classify viral reads and had comparable sensitivity and specificity to slower full read alignment approaches, such as BLASTn, for the data sets examined.
在疫苗和基于生物技术的药物生产过程中,检测外来病原体对于确保最终产品不受任何潜在病毒污染至关重要。提高病毒检测的速度和准确性有助于加速开发时间表,并确保患者安全。在这里,几种快速病毒宏基因组学方法在模拟下一代测序 (NGS) 数据集和来自 CHO-K1 和 HeLa 细胞系中进行的病毒掺入研究的现有数据集上进行了测试。观察到这些快速方法对于这些数据集的 NGS 病毒检测的全读对齐方法具有可比的灵敏度,但它们的特异性可以提高。建议使用 KrakenUniq 首先过滤宿主读取,然后根据剩余读取的数量选择病毒分类工具的方法作为测试中检测非潜伏和非内源性病毒的首选方法。这种方法对于检查的数据集显示出合理的灵敏度和特异性,并且需要的时间和内存比全读对齐方法少。下一代测序 (NGS) 已被提议作为一种补充方法,用于检测生物疗法和疫苗生产中的外来病毒,以替代当前的 和 方法。在 NGS 可以作为主要病毒检测技术在工业中建立之前,需要进一步研究识别和分类病毒 NGS 读取所需的各个生物信息学分析方面。在这项研究中,测试并比较了快速宏基因组学工具在生物制药相关样本中检测病毒的能力,以推荐一种有效的方法。结果表明,KrakenUniq 可以快速准确地过滤宿主序列并分类病毒读取,并且对于检查的数据集,其灵敏度和特异性与较慢的全读对齐方法(如 BLASTn)相当。