Departments of Physiology and Cell Biology, The Ohio State University Wexner Medical Center, Columbus, OH, USA.
Dorothy M. Davis Heart and Lung Research Institute, The Ohio State University Wexner Medical Center, Columbus, OH, USA.
Acta Physiol (Oxf). 2021 Jul;232(3):e13666. doi: 10.1111/apha.13666. Epub 2021 Apr 29.
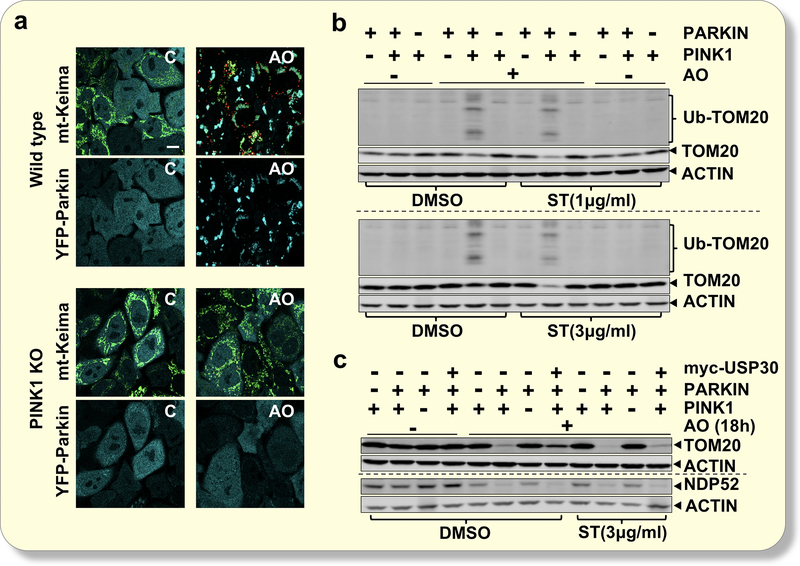
Mitophagy is the regulated process that targets damaged or dysfunctional mitochondria for lysosomal-mediated removal. This process is an essential element of mitochondrial quality control, and dysregulation of mitophagy may contribute to a host of diseases, most notably neurodegenerative conditions such as Parkinson's disease. Mitochondria targeted for mitophagic destruction are molecularly marked by the ubiquitination of several outer mitochondrial membrane (OMM) proteins. This ubiquitination is positively regulated, in part, by the mitochondrial-targeted kinase PINK1 and the E3 ubiquitin ligase Parkin. In contrast, the reverse phenomenon, deubiquitination, removes ubiquitin from Parkin substrates embedded in the OMM proteins, antagonizing mitophagy. Recent evidence suggests that the mitochondrial deubiquitinase USP30 negatively regulates Parkin-mediated mitophagy, providing opportunities to identify USP30 inhibitors and test for their effects in augmenting mitophagy. Here we will characterize a USP30 inhibitor and demonstrate how the pharmacological inhibition of USP30 can augment stress-induced mitophagic flux.
We have conducted mitophagy and mitochondrial analyses in cultured cells. We have determined the plasma pharmacokinetics of the USP30 inhibitor in mice and conducted analyses using the mt-Keima mice to measure in vivo mitophagy directly.
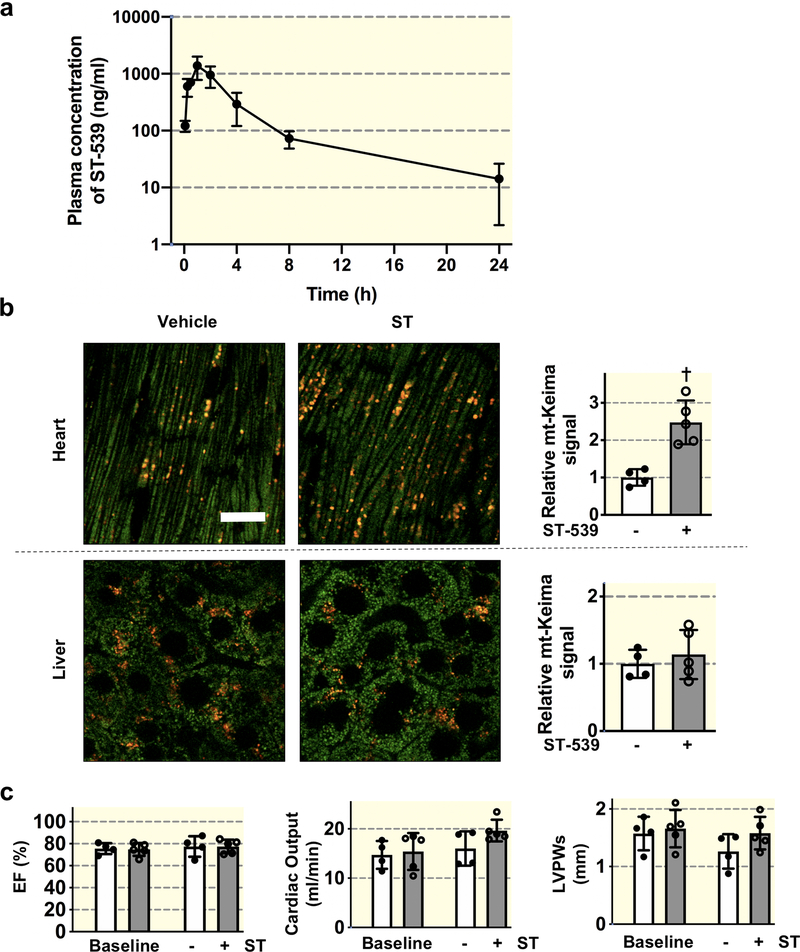
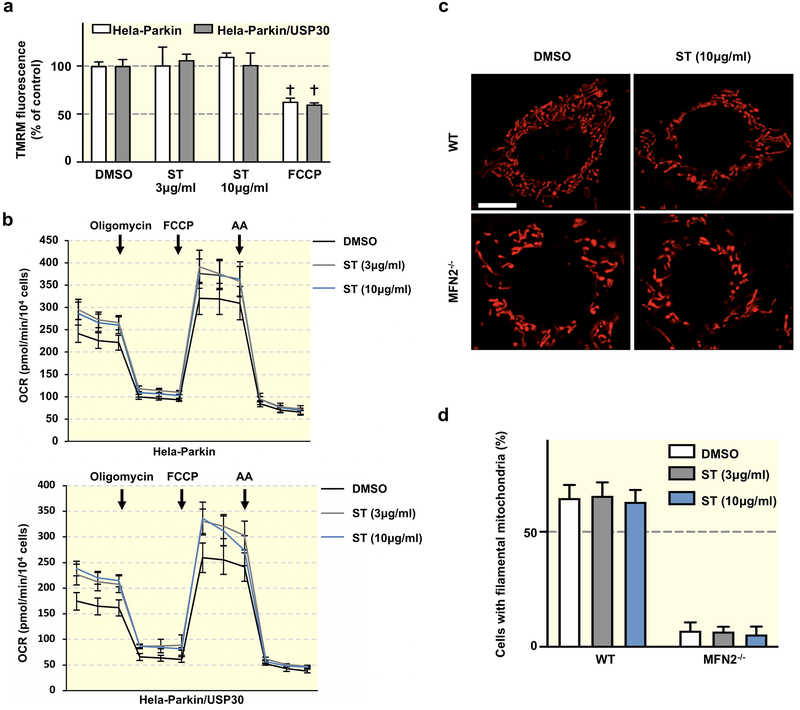
The compound has minimal mitochondrial toxicity in cultured cells and is tolerated well in mice. Interestingly, we demonstrated tissue-specific induction of mitophagy following USP30 pharmacological inhibition. In particular, pharmacological inhibition of USP30 induces a significant increase in cardiac mitophagy without detriment to cardiac function.
Our data support the evidence that USP30 inhibition may serve as a specific strategy to selectively increase mitophagic flux, allowing for the development of novel therapeutic approaches.
自噬是一种靶向受损或功能失调线粒体并通过溶酶体介导清除的过程。这个过程是线粒体质量控制的一个重要组成部分,自噬的失调可能导致多种疾病,尤其是帕金森病等神经退行性疾病。被靶向进行自噬破坏的线粒体通过几种外膜(OMM)蛋白的泛素化来进行分子标记。这种泛素化在一定程度上受到线粒体靶向激酶 PINK1 和 E3 泛素连接酶 Parkin 的正向调节。相比之下,相反的现象,即去泛素化,会从嵌入 OMM 蛋白中的 Parkin 底物上去除泛素,从而拮抗自噬。最近的证据表明,线粒体去泛素酶 USP30 负向调节 Parkin 介导的自噬,为鉴定 USP30 抑制剂并测试其增强自噬的效果提供了机会。在这里,我们将对 USP30 抑制剂进行表征,并展示如何通过药理学抑制 USP30 来增强应激诱导的自噬通量。
我们在培养的细胞中进行了自噬和线粒体分析。我们已经确定了 USP30 抑制剂在小鼠中的血浆药代动力学,并使用 mt-Keima 小鼠进行了分析,以直接测量体内自噬。
该化合物在培养细胞中对线粒体毒性极小,在小鼠中耐受性良好。有趣的是,我们证明了 USP30 药理学抑制后会导致组织特异性的自噬诱导。特别是,USP30 的药理学抑制会显著增加心脏自噬,而不会损害心脏功能。
我们的数据支持了 USP30 抑制可能作为一种选择性增加自噬通量的特定策略的证据,为开发新的治疗方法提供了可能。