European Molecular Biology Laboratory, EMBL-European Bioinformatics Institute (EMBL-EBI), Hinxton, Cambridge, CB10 1SD, UK.
Department of Informatics, University of Bergen, 5020, Bergen, Norway.
Sci Data. 2021 Apr 23;8(1):115. doi: 10.1038/s41597-021-00890-2.
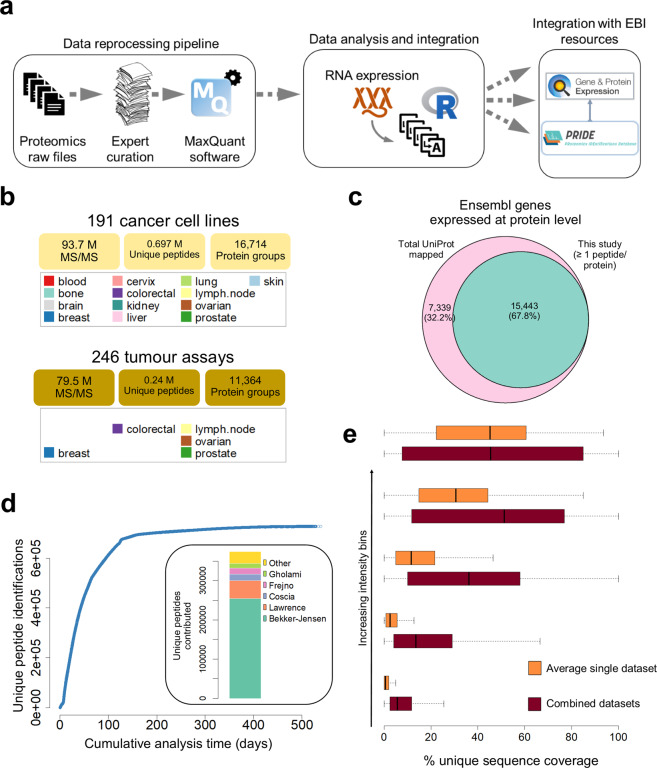
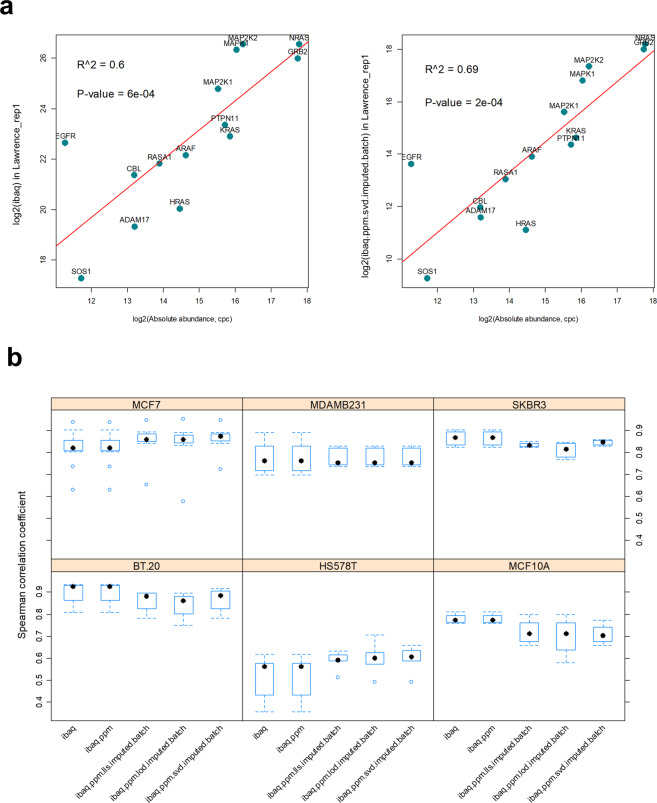
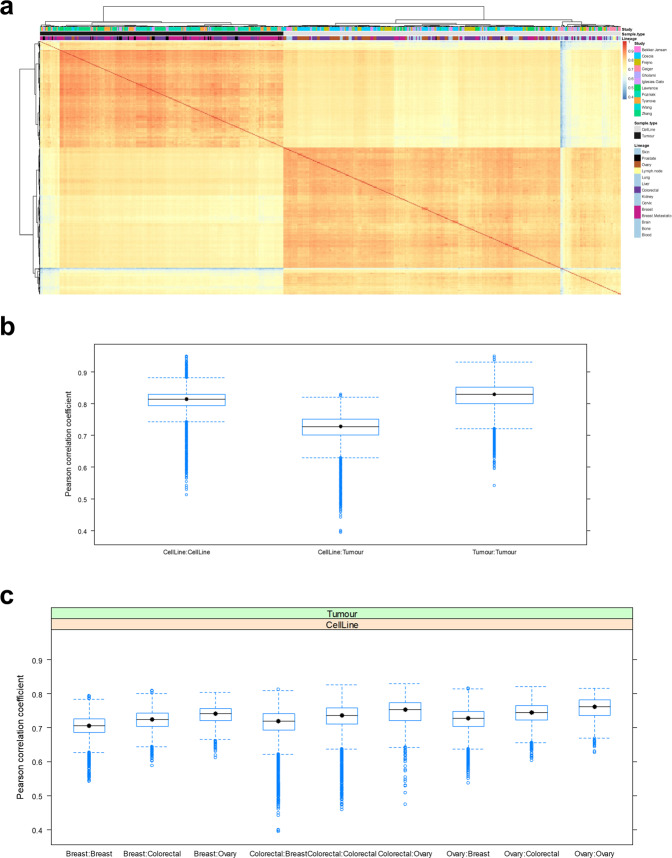
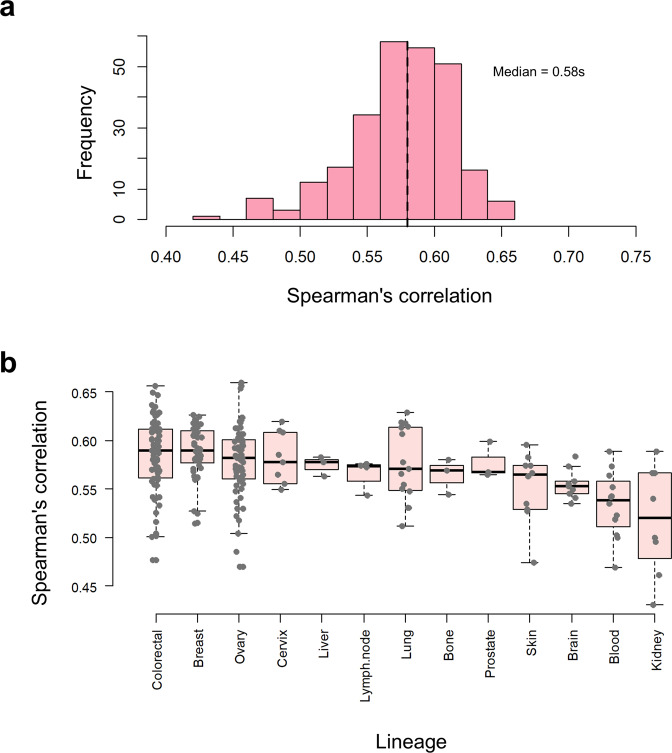
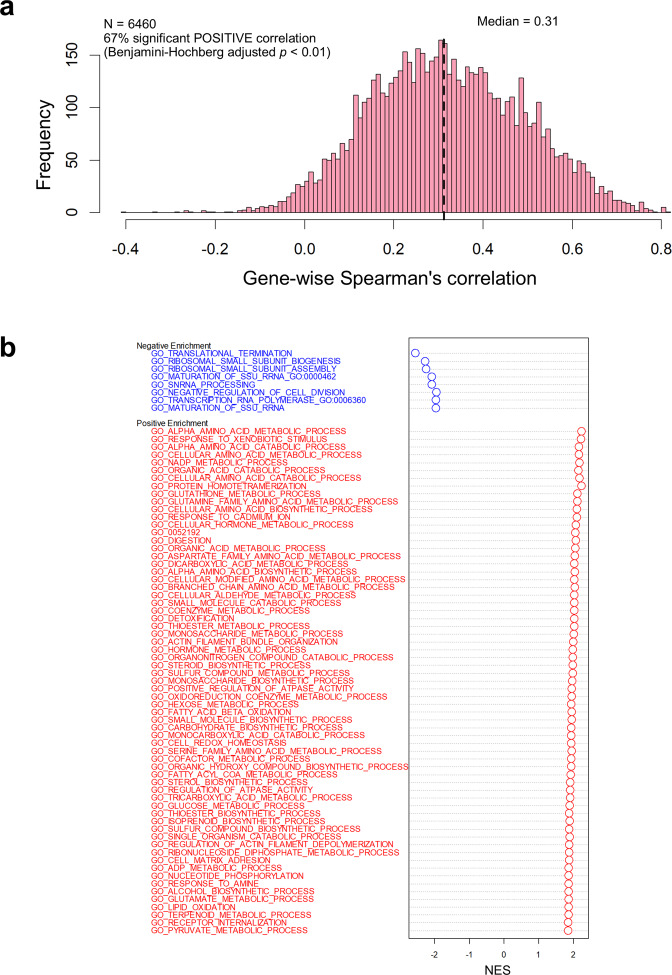
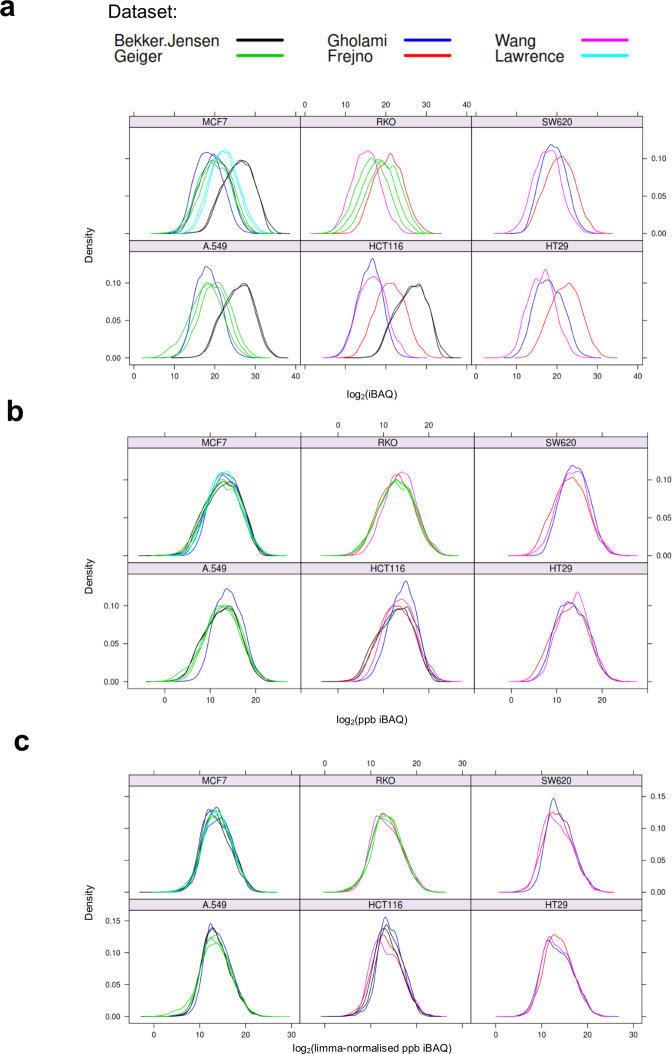
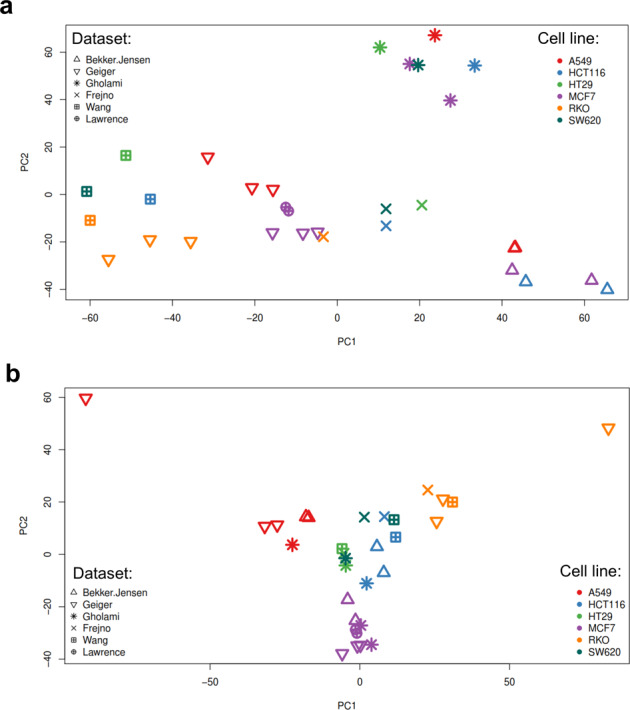
Using 11 proteomics datasets, mostly available through the PRIDE database, we assembled a reference expression map for 191 cancer cell lines and 246 clinical tumour samples, across 13 lineages. We found unique peptides identified only in tumour samples despite a much higher coverage in cell lines. These were mainly mapped to proteins related to regulation of signalling receptor activity. Correlations between baseline expression in cell lines and tumours were calculated. We found these to be highly similar across all samples with most similarity found within a given sample type. Integration of proteomics and transcriptomics data showed median correlation across cell lines to be 0.58 (range between 0.43 and 0.66). Additionally, in agreement with previous studies, variation in mRNA levels was often a poor predictor of changes in protein abundance. To our knowledge, this work constitutes the first meta-analysis focusing on cancer-related public proteomics datasets. We therefore also highlight shortcomings and limitations of such studies. All data is available through PRIDE dataset identifier PXD013455 and in Expression Atlas.
使用 11 个蛋白质组学数据集,这些数据集主要通过 PRIDE 数据库获得,我们为 191 种癌细胞系和 246 个临床肿瘤样本组装了一个参考表达图谱,涵盖了 13 个谱系。我们发现了仅在肿瘤样本中识别出的独特肽,尽管在细胞系中覆盖度更高。这些主要映射到与信号受体活性调节相关的蛋白质。计算了细胞系和肿瘤中基线表达之间的相关性。我们发现这些在所有样本中非常相似,大多数相似性在给定的样本类型内发现。蛋白质组学和转录组学数据的整合显示,细胞系之间的中位数相关性为 0.58(范围在 0.43 到 0.66 之间)。此外,与之前的研究一致,mRNA 水平的变化通常不能很好地预测蛋白质丰度的变化。据我们所知,这项工作是首次针对与癌症相关的公共蛋白质组学数据集进行的荟萃分析。因此,我们还强调了此类研究的缺点和局限性。所有数据均可通过 PRIDE 数据集标识符 PXD013455 和 Expression Atlas 获取。