Akbani Rehan, Ng Patrick Kwok Shing, Werner Henrica M J, Shahmoradgoli Maria, Zhang Fan, Ju Zhenlin, Liu Wenbin, Yang Ji-Yeon, Yoshihara Kosuke, Li Jun, Ling Shiyun, Seviour Elena G, Ram Prahlad T, Minna John D, Diao Lixia, Tong Pan, Heymach John V, Hill Steven M, Dondelinger Frank, Städler Nicolas, Byers Lauren A, Meric-Bernstam Funda, Weinstein John N, Broom Bradley M, Verhaak Roeland G W, Liang Han, Mukherjee Sach, Lu Yiling, Mills Gordon B
1] Department of Bioinformatics and Computational Biology, 1400 Pressler St., The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA [2].
1] Department of Systems Biology, 1515 Holcombe Blvd, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA [2].
Nat Commun. 2014 May 29;5:3887. doi: 10.1038/ncomms4887.
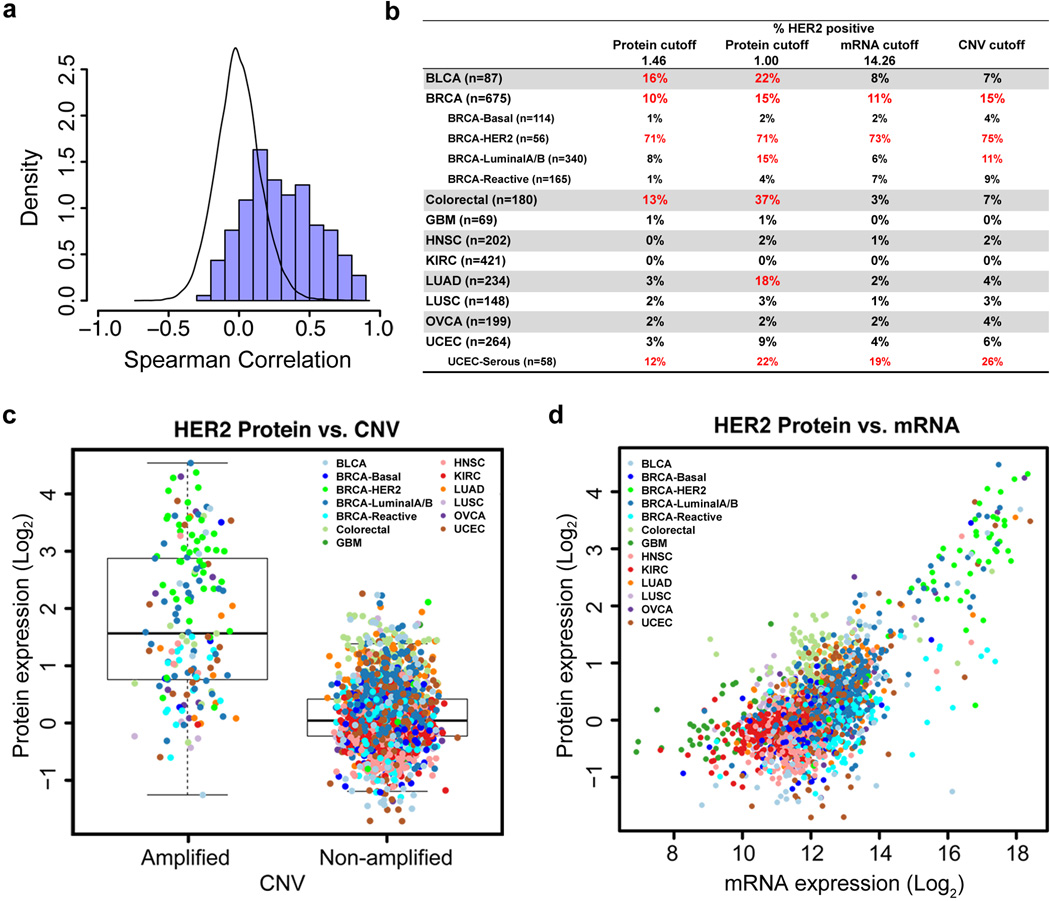
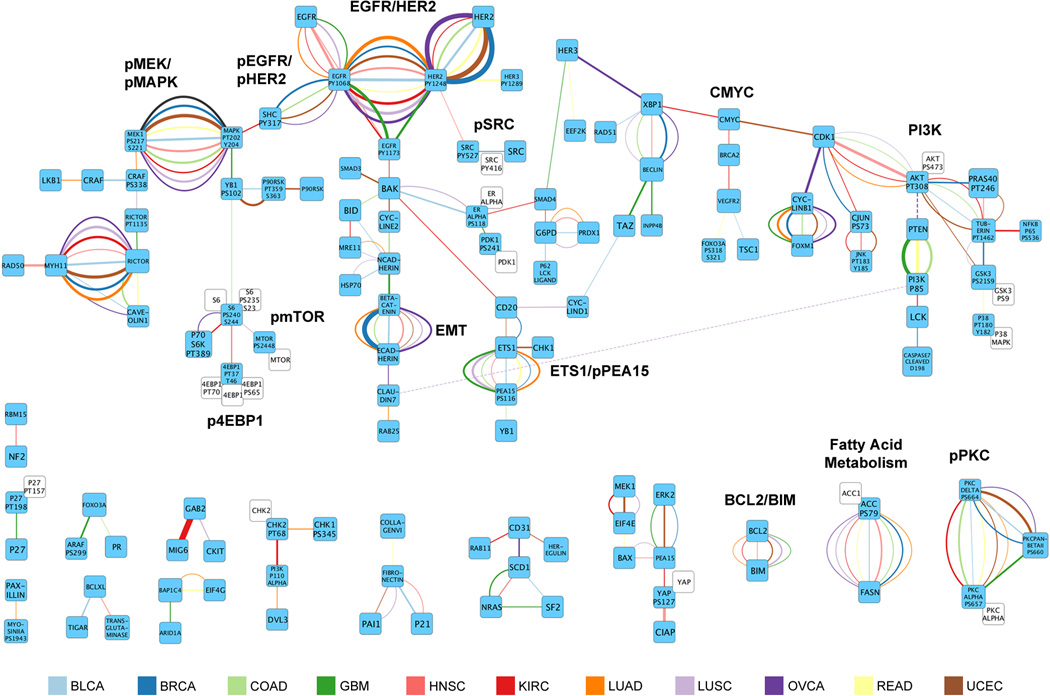
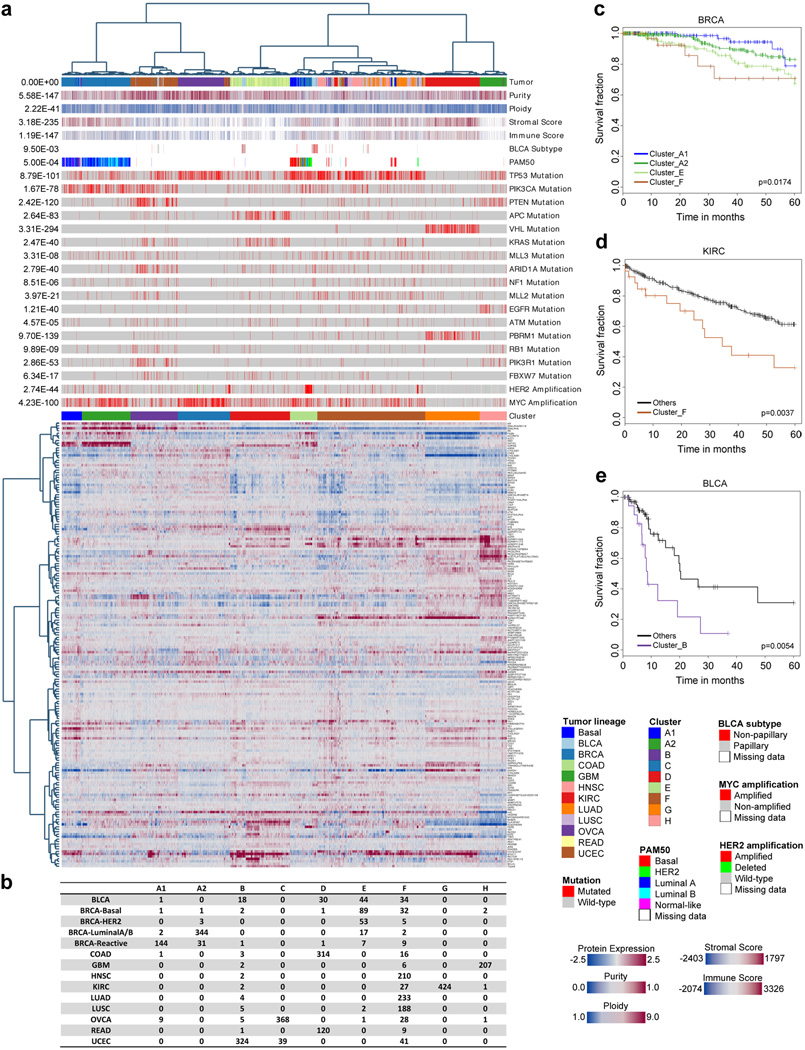
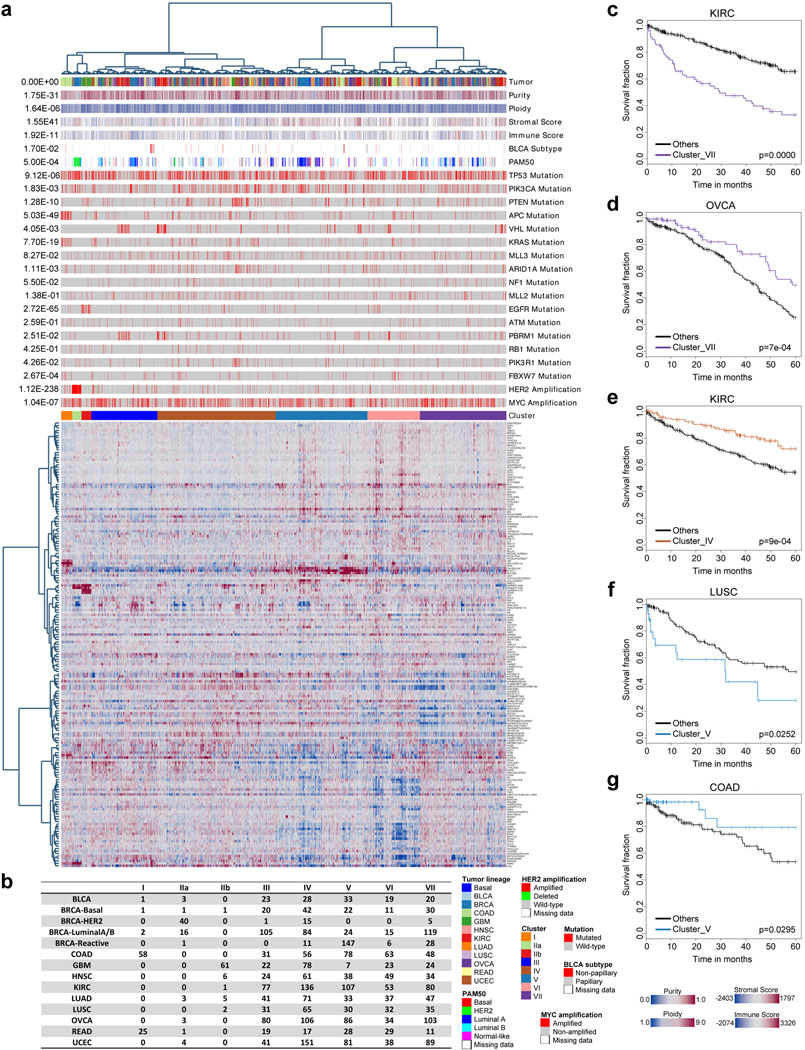
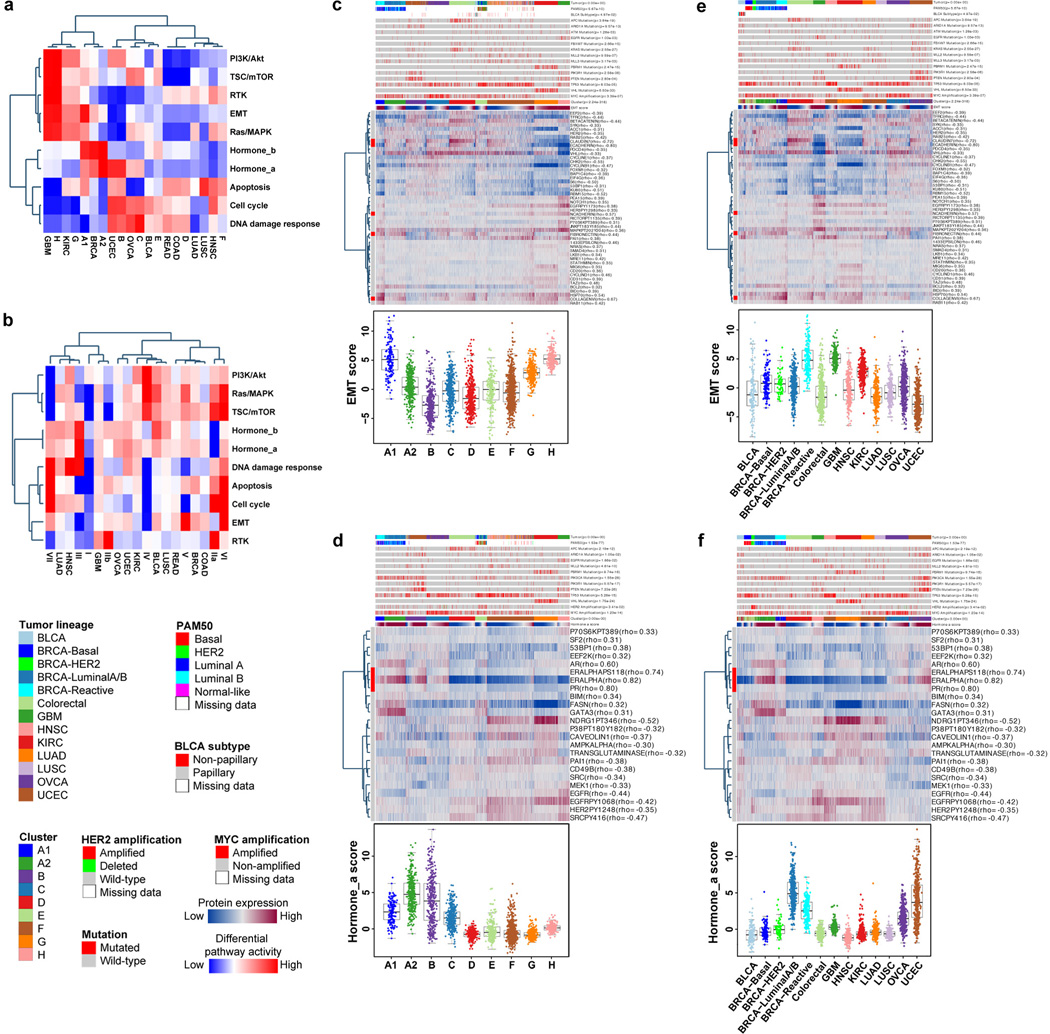
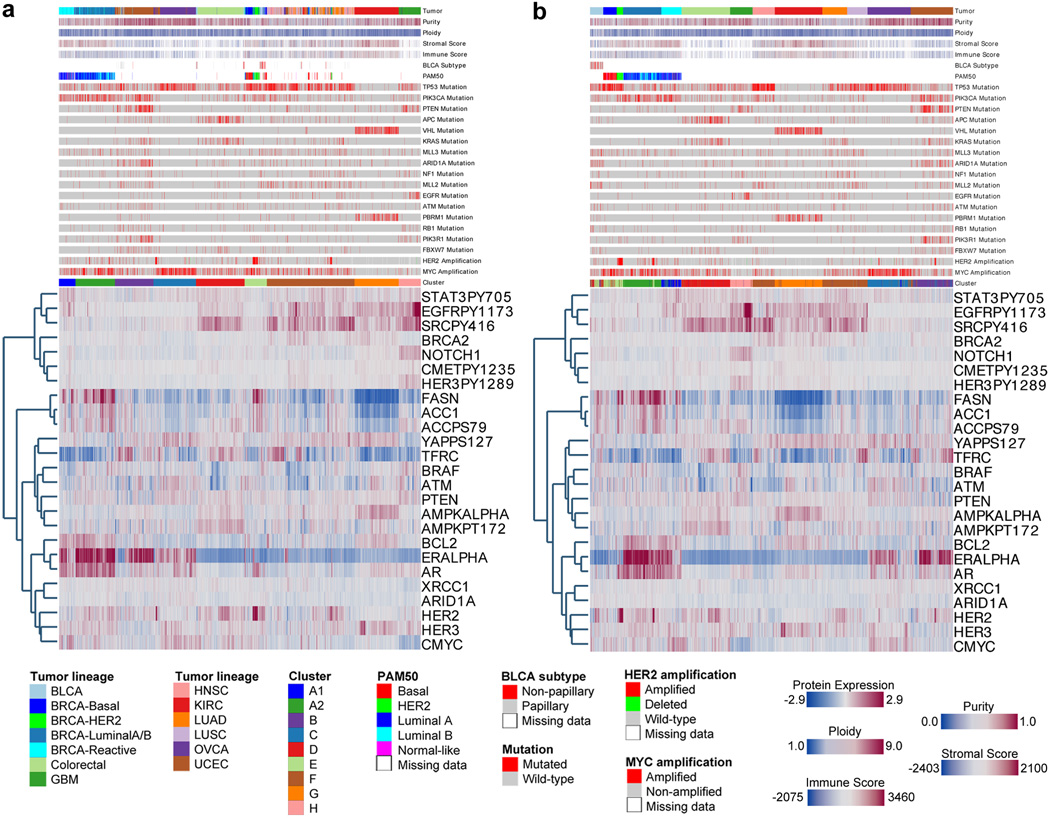
Protein levels and function are poorly predicted by genomic and transcriptomic analysis of patient tumours. Therefore, direct study of the functional proteome has the potential to provide a wealth of information that complements and extends genomic, epigenomic and transcriptomic analysis in The Cancer Genome Atlas (TCGA) projects. Here we use reverse-phase protein arrays to analyse 3,467 patient samples from 11 TCGA 'Pan-Cancer' diseases, using 181 high-quality antibodies that target 128 total proteins and 53 post-translationally modified proteins. The resultant proteomic data are integrated with genomic and transcriptomic analyses of the same samples to identify commonalities, differences, emergent pathways and network biology within and across tumour lineages. In addition, tissue-specific signals are reduced computationally to enhance biomarker and target discovery spanning multiple tumour lineages. This integrative analysis, with an emphasis on pathways and potentially actionable proteins, provides a framework for determining the prognostic, predictive and therapeutic relevance of the functional proteome.
通过对患者肿瘤进行基因组和转录组分析,很难预测蛋白质水平和功能。因此,对功能蛋白质组进行直接研究有可能提供大量信息,补充并扩展癌症基因组图谱(TCGA)项目中的基因组、表观基因组和转录组分析。在此,我们使用反相蛋白质阵列,利用181种针对128种总蛋白和53种翻译后修饰蛋白的高质量抗体,分析来自11种TCGA“泛癌”疾病的3467份患者样本。将所得蛋白质组数据与相同样本的基因组和转录组分析相结合,以识别肿瘤谱系内部和之间的共性、差异、新出现的通路和网络生物学。此外,通过计算减少组织特异性信号,以加强跨多个肿瘤谱系的生物标志物和靶点发现。这种综合分析,重点关注通路和潜在的可作用蛋白,为确定功能蛋白质组的预后、预测和治疗相关性提供了一个框架。