Chemical and Biological Crystallography, Department of Chemistry, School of Natural Sciences, Shiv Nadar University, Dadri 201314, Uttar Pradesh, India.
Molecules. 2021 Apr 29;26(9):2605. doi: 10.3390/molecules26092605.
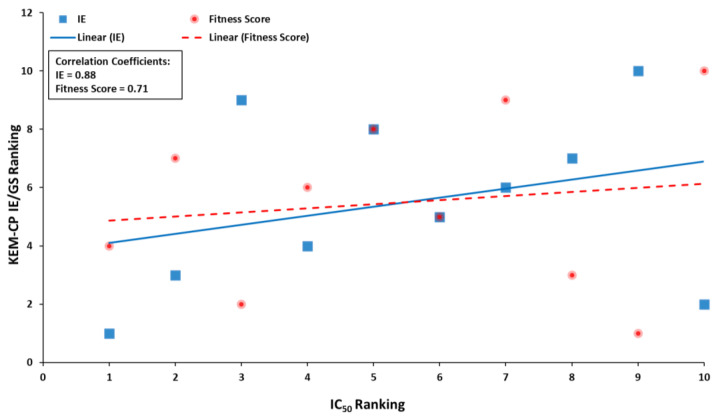
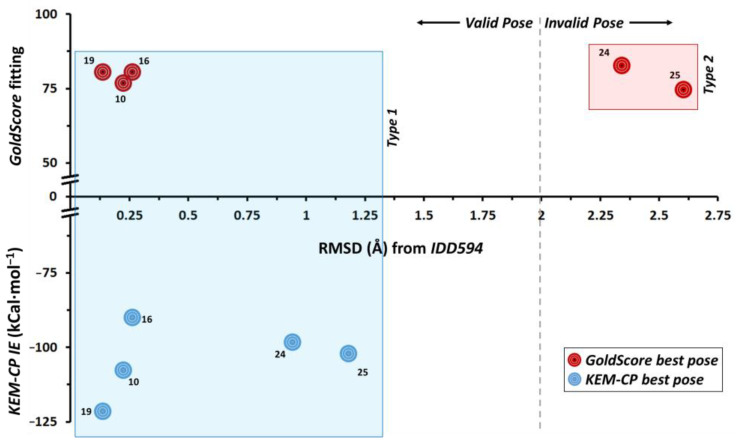
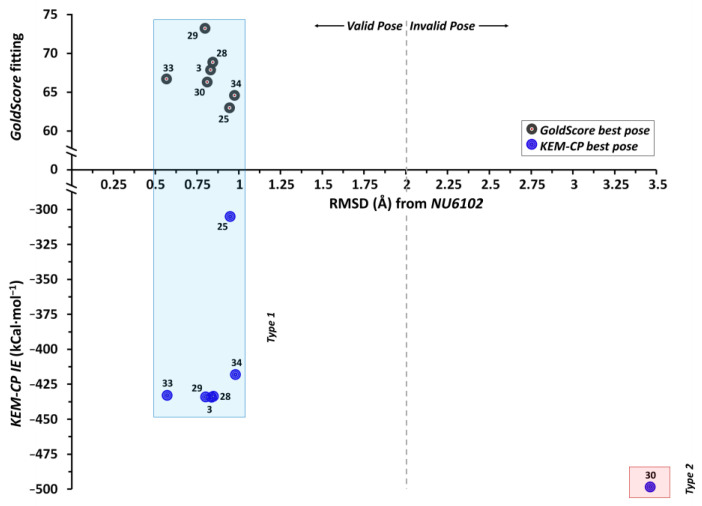
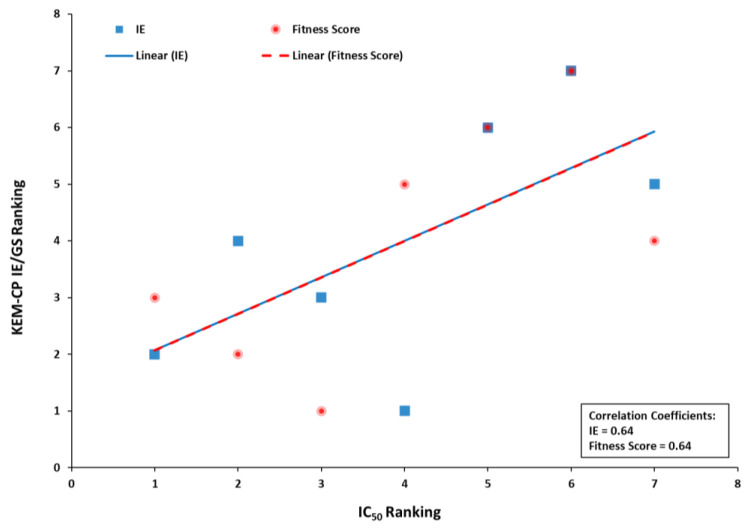
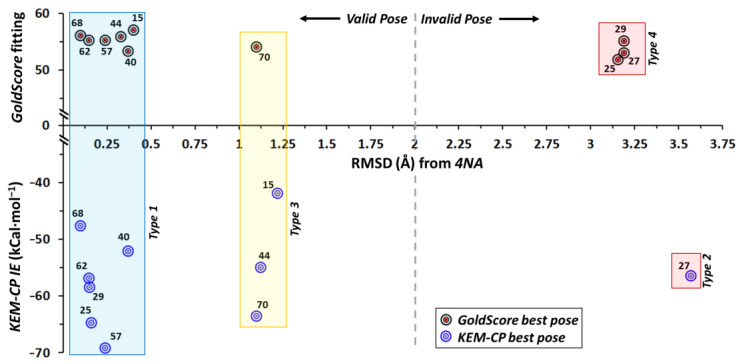
Optimization of lead structures is crucial for drug discovery. However, the accuracy of such a prediction using the traditional molecular docking approach remains a major concern. Our study demonstrates that the employment of quantum crystallographic approach-counterpoise corrected kernel energy method (KEM-CP) can improve the accuracy by and large. We select human aldose reductase at 0.66 Å, cyclin dependent kinase 2 at 2.0 Å and estrogen receptor β at 2.7 Å resolutions with active site environment ranging from highly hydrophilic to moderate to highly hydrophobic and several of their known ligands. Overall, the use of KEM-CP alongside the GoldScore resulted superior prediction than the GoldScore alone. Unlike GoldScore, the KEM-CP approach is neither environment-specific nor structural resolution dependent, which highlights its versatility. Further, the ranking of the ligands based on the KEM-CP results correlated well with that of the experimental IC values. This computationally inexpensive yet simple approach is expected to ease the process of virtual screening of potent ligands, and it would advance the drug discovery research.
先导结构的优化对于药物发现至关重要。然而,传统分子对接方法在进行此类预测时的准确性仍然是一个主要关注点。我们的研究表明,采用量子晶体学方法——校正后对势能(CP)的内核能量法(KEM-CP)可以在很大程度上提高准确性。我们选择了 0.66Å 的人醛糖还原酶、2.0Å 的细胞周期蛋白依赖性激酶 2 和 2.7Å 的雌激素受体β,并对其活性部位环境进行了分类,从高度亲水到中度亲脂再到高度亲脂,以及它们的几种已知配体。总体而言,与仅使用 GoldScore 相比,使用 KEM-CP 和 GoldScore 的组合具有更好的预测性能。与 GoldScore 不同,KEM-CP 方法既不是环境特异性的,也不是结构分辨率依赖的,这突出了其通用性。此外,根据 KEM-CP 结果对配体进行的排序与实验 IC 值的排序非常吻合。这种计算成本低廉但简单的方法有望简化潜在配体的虚拟筛选过程,并推进药物发现研究。