Echave Julian
Instituto de Ciencias Físicas, Escuela de Ciencia y Tecnología, Universidad Nacional de San Martín, San Martín, Buenos Aires, Argentina.
PeerJ. 2021 Apr 21;9:e11330. doi: 10.7717/peerj.11330. eCollection 2021.
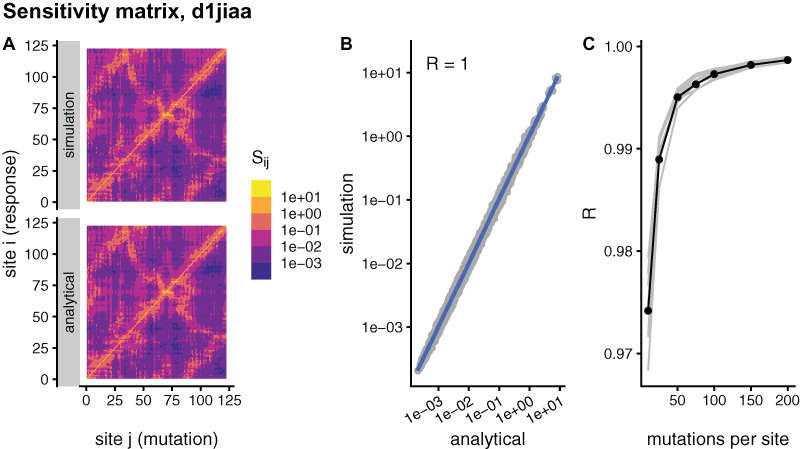
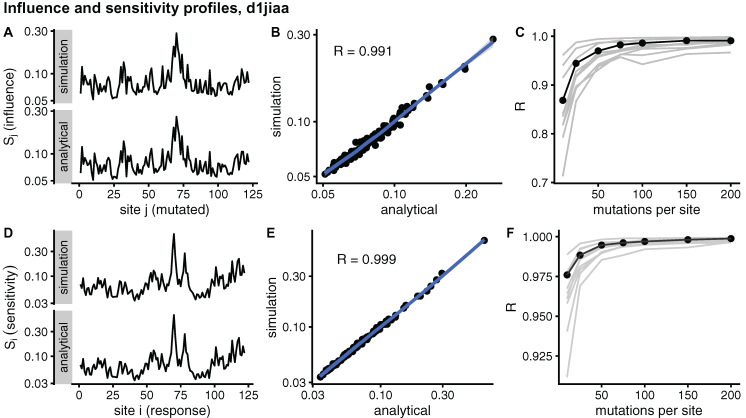
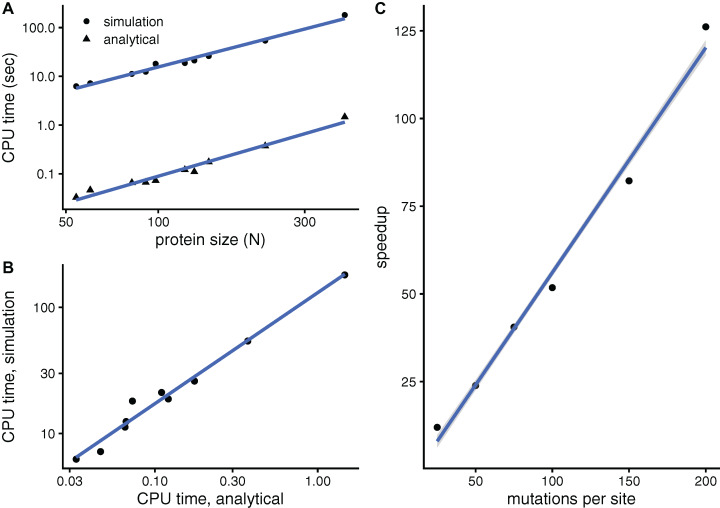
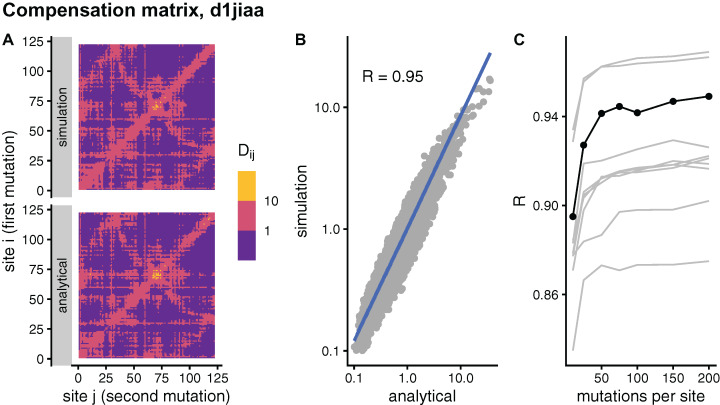
Studying the effect of perturbations on protein structure is a basic approach in protein research. Important problems, such as predicting pathological mutations and understanding patterns of structural evolution, have been addressed by computational simulations that model mutations using forces and predict the resulting deformations. In single mutation-response scanning simulations, a sensitivity matrix is obtained by averaging deformations over point mutations. In double mutation-response scanning simulations, a compensation matrix is obtained by minimizing deformations over pairs of mutations. These very useful simulation-based methods may be too slow to deal with large proteins, protein complexes, or large protein databases. To address this issue, I derived analytical closed formulas to calculate the sensitivity and compensation matrices directly, without simulations. Here, I present these derivations and show that the resulting analytical methods are much faster than their simulation counterparts.
研究扰动对蛋白质结构的影响是蛋白质研究中的一种基本方法。诸如预测病理突变和理解结构进化模式等重要问题,已通过计算模拟得到解决,这些模拟使用力对突变进行建模并预测由此产生的变形。在单突变响应扫描模拟中,通过对单点突变的变形进行平均来获得灵敏度矩阵。在双突变响应扫描模拟中,通过最小化成对突变的变形来获得补偿矩阵。这些基于模拟的非常有用的方法可能处理大型蛋白质、蛋白质复合物或大型蛋白质数据库时速度太慢。为了解决这个问题,我推导了分析性的封闭公式,无需模拟即可直接计算灵敏度和补偿矩阵。在此,我展示这些推导过程,并表明由此产生的分析方法比其模拟对应方法快得多。