Interuniversity Institute of Bioinformatics in Brussels, ULB-VUB, La Plaine Campus, Triomflaan, 1050, Brussels, Belgium.
ESAT-STADIUS, KU Leuven, Kasteelpark Arenberg 10, 3001, Leuven, Belgium.
Sci Rep. 2018 Nov 19;8(1):16980. doi: 10.1038/s41598-018-34959-7.
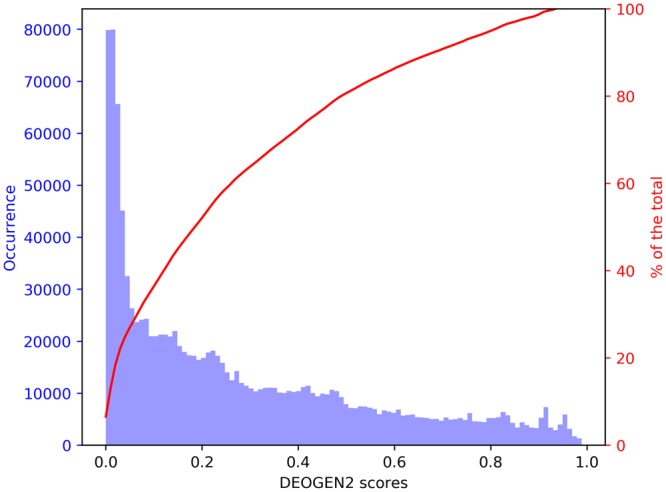
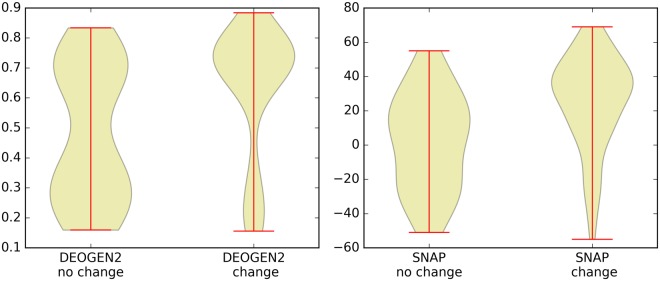
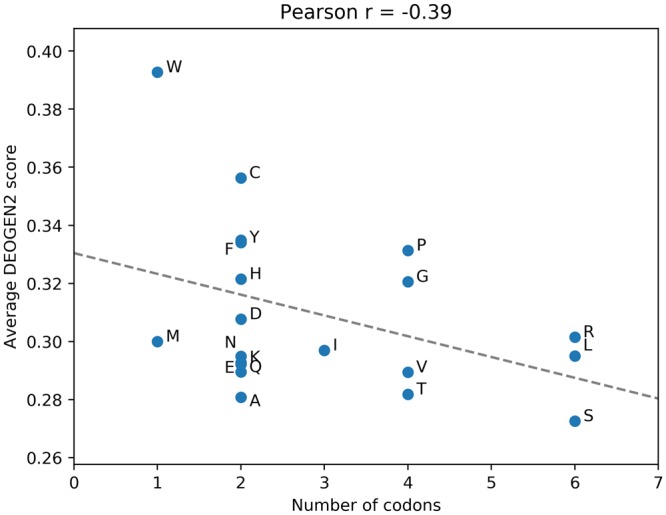
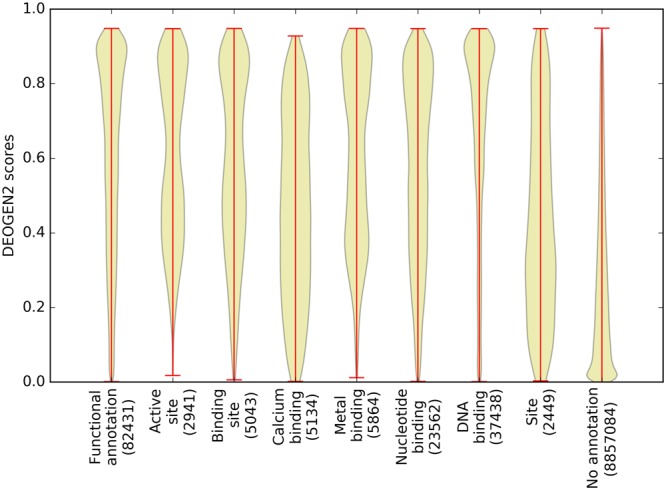
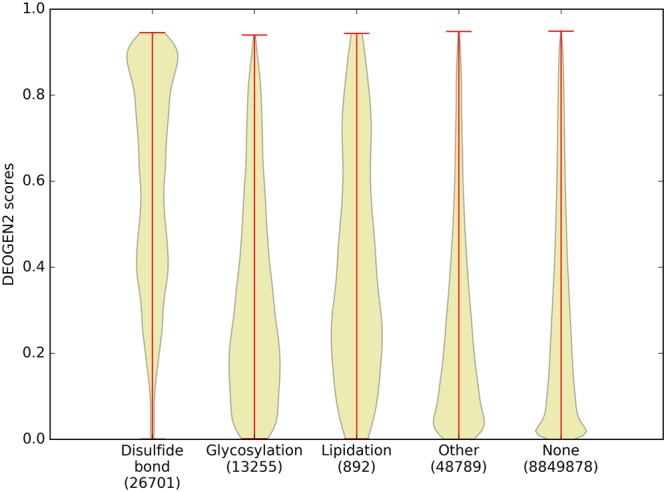
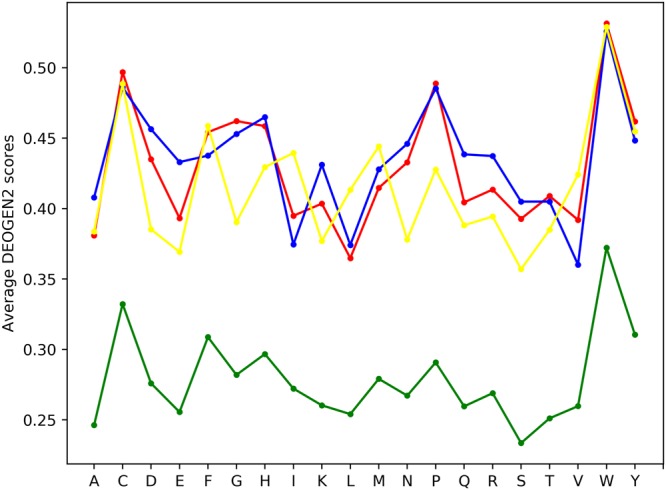
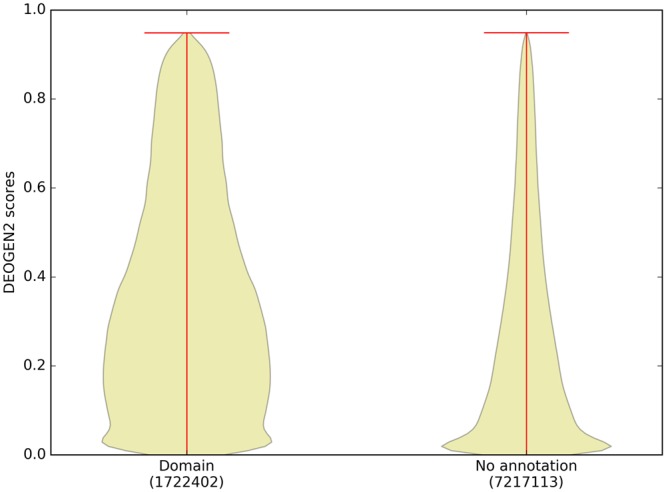
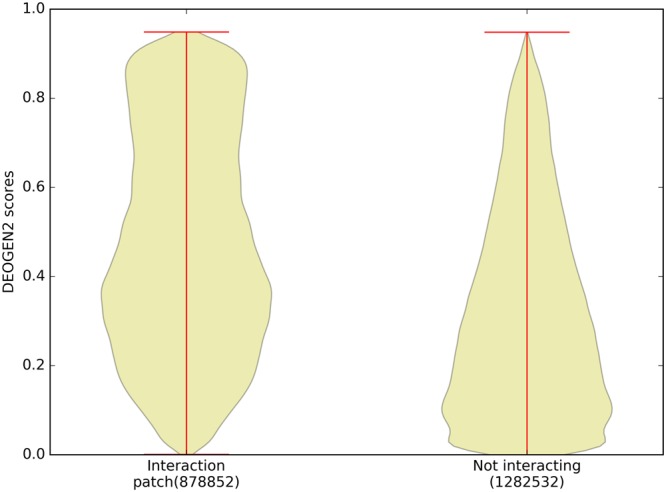
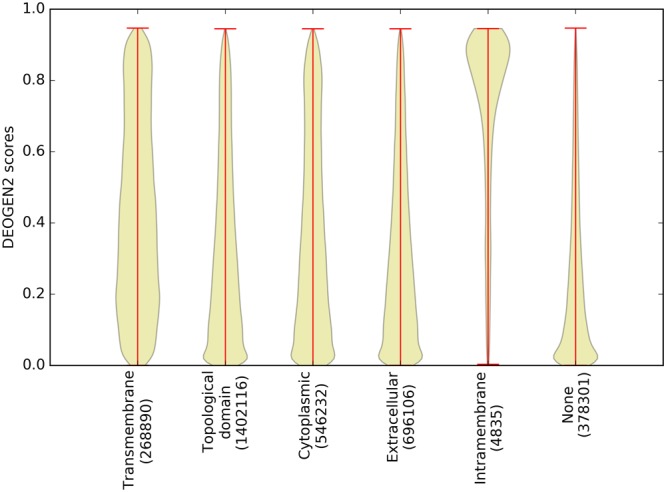
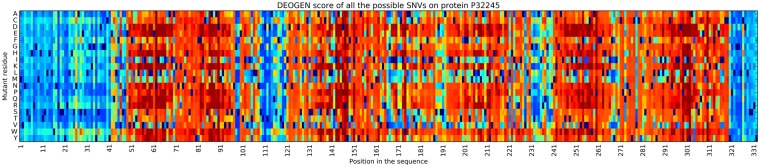
Next generation sequencing technologies are providing increasing amounts of sequencing data, paving the way for improvements in clinical genetics and precision medicine. The interpretation of the observed genomic variants in the light of their phenotypic effects is thus emerging as a crucial task to solve in order to advance our understanding of how exomic variants affect proteins and how the proteins' functional changes affect human health. Since the experimental evaluation of the effects of every observed variant is unfeasible, Bioinformatics methods are being developed to address this challenge in-silico, by predicting the impact of millions of variants, thus providing insight into the deleteriousness landscape of entire proteomes. Here we show the feasibility of this approach by using the recently developed DEOGEN2 variant-effect predictor to perform the largest in-silico mutagenesis scan to date. We computed the deleteriousness score of 170 million variants over 15000 human proteins and we analysed the results, investigating how the predicted deleteriousness landscape of the proteins relates to known functionally and structurally relevant protein regions and biophysical properties. Moreover, we qualitatively validated our results by comparing them with two mutagenesis studies targeting two specific proteins, showing the consistency of DEOGEN2 predictions with respect to experimental data.
下一代测序技术正在提供越来越多的测序数据,为临床遗传学和精准医学的发展铺平了道路。因此,如何根据表型效应来解释观察到的基因组变异,已成为解决这一问题的关键任务,这有助于我们了解外显子变异如何影响蛋白质,以及蛋白质功能变化如何影响人类健康。由于对每个观察到的变异的实验评估是不可行的,因此正在开发生物信息学方法来通过预测数百万个变异的影响来解决这个问题,从而深入了解整个蛋白质组的有害性景观。在这里,我们通过使用最近开发的 DEOGEN2 变体效应预测器来展示这种方法的可行性,进行了迄今为止最大的基于计算的诱变扫描。我们计算了 1.7 亿个变体在 15000 个人类蛋白质上的有害性评分,并对结果进行了分析,研究了蛋白质的预测有害性景观与已知功能和结构相关的蛋白质区域和生物物理性质之间的关系。此外,我们通过将结果与针对两个特定蛋白质的两个诱变研究进行定性比较来验证我们的结果,表明 DEOGEN2 预测与实验数据的一致性。