Division of Endocrinology and Metabolism, Department of Internal Medicine, Jichi Medical University.
Department of Diabetes and Metabolic Diseases, Graduate School of Medicine, The University of Tokyo.
J Atheroscler Thromb. 2021 Oct 1;28(10):1009-1019. doi: 10.5551/jat.RV17056. Epub 2021 May 16.
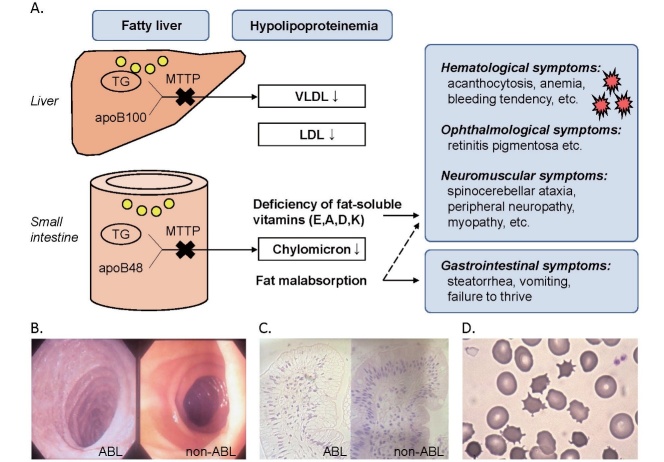
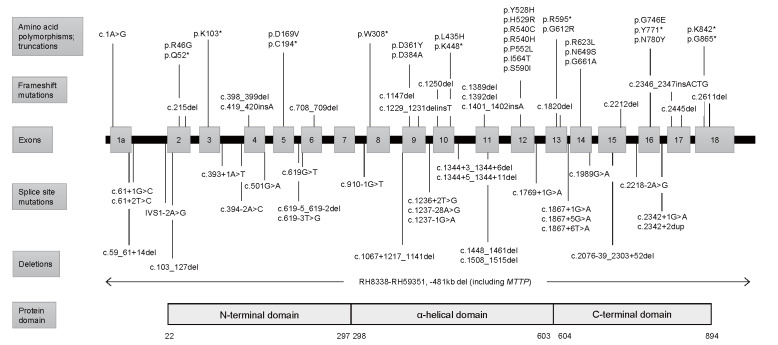
Abetalipoproteinemia (ABL) is a rare autosomal recessive disorder caused by biallelic pathogenic mutations in the MTTP gene. Deficiency of microsomal triglyceride transfer protein (MTTP) abrogates the assembly of apolipoprotein (apo) B-containing lipoprotein in the intestine and liver, resulting in malabsorption of fat and fat-soluble vitamins and severe hypolipidemia. Patients with ABL typically manifest steatorrhea, vomiting, and failure to thrive in infancy. The deficiency of fat-soluble vitamins progressively develops into a variety of symptoms later in life, including hematological (acanthocytosis, anemia, bleeding tendency, etc.), neuromuscular (spinocerebellar ataxia, peripheral neuropathy, myopathy, etc.), and ophthalmological symptoms (e.g., retinitis pigmentosa). If left untreated, the disease can be debilitating and even lethal by the third decade of life due to the development of severe complications, such as blindness, neuromyopathy, and respiratory failure. High dose vitamin supplementation is the mainstay for treatment and may prevent, delay, or alleviate the complications and improve the prognosis, enabling some patients to live to the eighth decade of life. However, it cannot fully prevent or restore impaired function. Novel therapeutic modalities that improve quality of life and prognosis are awaited. The aim of this review is to 1) summarize the pathogenesis, clinical signs and symptoms, diagnosis, and management of ABL, and 2) propose diagnostic criteria that define eligibility to receive financial support from the Japanese government for patients with ABL as a rare and intractable disease. In addition, our diagnostic criteria and the entry criterion of low-density lipoprotein cholesterol (LDL-C) <15 mg/dL and apoB <15 mg/dL can be useful in universal or opportunistic screening for the disease. Registry research on ABL is currently ongoing to better understand the disease burden and unmet needs of this life-threatening disease with few therapeutic options.
β-脂蛋白缺乏症(ABL)是一种罕见的常染色体隐性遗传病,由 MTTP 基因的双等位致病性突变引起。微粒体甘油三酯转移蛋白(MTTP)的缺乏会破坏肠和肝内载脂蛋白(apo)B 所含脂蛋白的组装,导致脂肪和脂溶性维生素吸收不良以及严重的低血脂症。ABL 患者通常在婴儿期表现出脂肪泻、呕吐和生长不良。脂溶性维生素的缺乏逐渐发展为以后生活中的各种症状,包括血液学(棘状红细胞增多症、贫血、出血倾向等)、神经肌肉(脊髓小脑共济失调、周围神经病、肌病等)和眼科症状(例如,视网膜色素变性)。如果不治疗,由于严重并发症的发展,如失明、神经肌肉病和呼吸衰竭,该疾病可能在生命的第三个十年致残甚至致命。高剂量维生素补充是治疗的主要方法,可以预防、延迟或减轻并发症并改善预后,使一些患者能够活到 80 岁。然而,它不能完全预防或恢复受损的功能。正在等待改善生活质量和预后的新型治疗方法。本综述的目的是:1)总结 ABL 的发病机制、临床体征和症状、诊断和管理,2)提出诊断标准,以确定 ABL 患者有资格获得日本政府作为罕见和难治性疾病的财政支持。此外,我们的诊断标准和 LDL-C<15mg/dL 和 apoB<15mg/dL 的切入点可以用于该疾病的普遍或机会性筛查。目前正在对 ABL 进行登记研究,以更好地了解这种危及生命的疾病的疾病负担和未满足的需求,该疾病的治疗选择很少。