Cassel Aliza, Rosenberg Nurit, Muhammad Emad, Livnat Tami, Dardik Rima, Berl Miriam, Preis Meir
Institute of Hematology Carmel Medical Center Haifa Israel.
The Israeli National Hemophilia Sheba Medical Center Tel Hashomer Israel.
Res Pract Thromb Haemost. 2021 Feb 25;5(4):e12407. doi: 10.1002/rth2.12407. eCollection 2021 May.
Measurement of factor VII (FVII) activity does not enable prediction of bleeding tendency in individuals with inherited FVII deficiency.
To characterize the molecular and functional features of FVII in a family with FVII deficiency and correlate them with the bleeding tendency.
PATIENTS/METHODS: We studied 7 family members with very low FVII activity using prothrombin time (PT), activated factor VII (FVIIa), FVII activity level, and thrombin generation. The factor 7 gene was sequenced and the mutation was analyzed by prediction software.
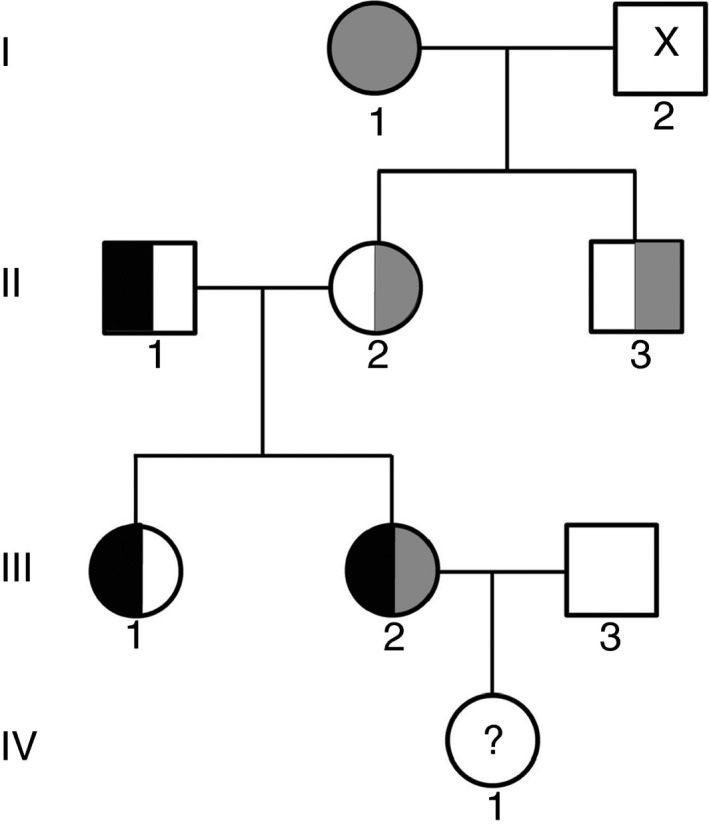
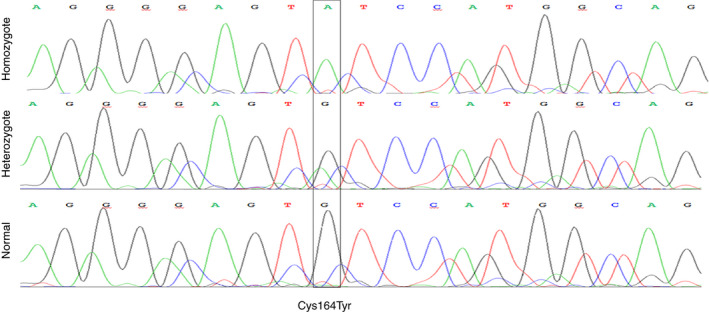
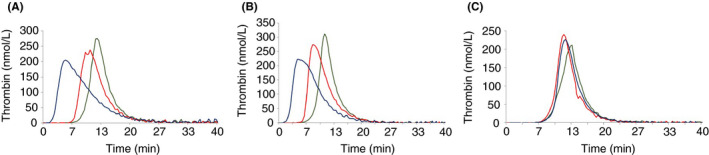
The proband has very low FVII activity (0%-4%), with PT ranging between 5% to 18% depending on the tissue factor (TF) origin. Direct sequencing demonstrated a single homozygous nucleotide substitution G > A in exon 6, predicting a novel missense mutation Cys164Tyr. Three members of the family were found to be heterozygous carriers of this mutation. One of them was a compound heterozygote, carrying both the Cys164Tyr and Ala244Val mutation (linked to Arg353Gln polymorphism). Her FVII activity and antigen levels were 3%-7% and 8%, respectively. The other heterozygous carriers demonstrated FVII activity of 41%-54%, FVII antigen of 46%-66%, and FVIIa activity of 30%. FVIIa was undetectable in the homozygous and compound heterozygous subjects. Thrombin generation was normal in the presence of calcium, but no response to TF addition was observed in the homozygous proband, and a reduced response was observed in the compound heterozygous subject.
The patient homozygous for the "Carmel" mutation has mild clinical manifestations despite very low FVII activity, which correlates with thrombin generation results.
因子VII(FVII)活性的测定无法预测遗传性FVII缺乏个体的出血倾向。
对一个FVII缺乏家族中FVII的分子和功能特征进行表征,并将其与出血倾向相关联。
患者/方法:我们使用凝血酶原时间(PT)、活化因子VII(FVIIa)、FVII活性水平和凝血酶生成,研究了7名FVII活性极低的家族成员。对因子7基因进行测序,并通过预测软件分析突变。
先证者的FVII活性极低(0%-4%),根据组织因子(TF)来源不同,PT在5%至18%之间。直接测序显示外显子6中存在单个纯合核苷酸替换G>A,预测为一个新的错义突变Cys164Tyr。该家族中有3名成员被发现是此突变的杂合携带者。其中一人为复合杂合子,同时携带Cys164Tyr和Ala244Val突变(与Arg353Gln多态性相关)。她的FVII活性和抗原水平分别为3%-7%和8%。其他杂合携带者的FVII活性为41%-54%,FVII抗原为46%-66%,FVIIa活性为30%。在纯合和复合杂合受试者中未检测到FVIIa。在有钙存在的情况下,凝血酶生成正常,但在纯合先证者中未观察到对添加TF的反应,在复合杂合受试者中观察到反应减弱。
尽管FVII活性极低,但“卡梅尔”突变纯合患者的临床表现较轻,这与凝血酶生成结果相关。