Department of Biological Sciences, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, United States.
J Chem Inf Model. 2021 Jun 28;61(6):2523-2529. doi: 10.1021/acs.jcim.1c00103. Epub 2021 May 24.
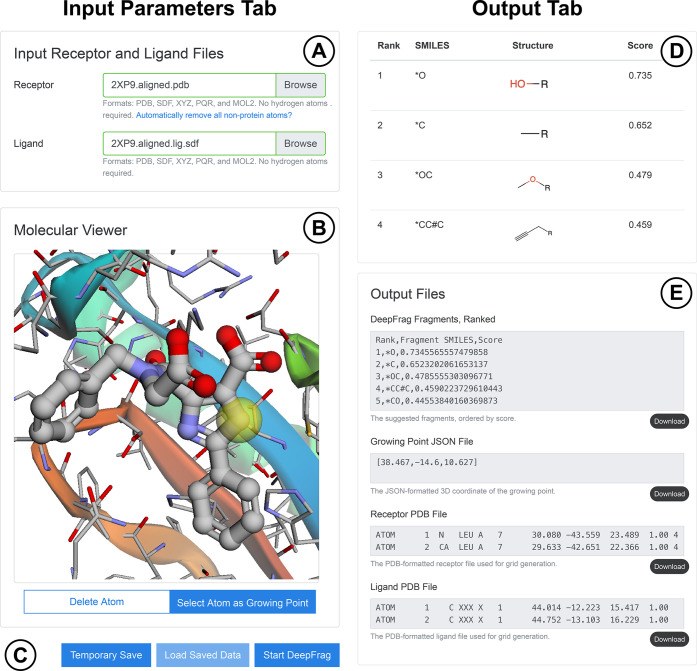
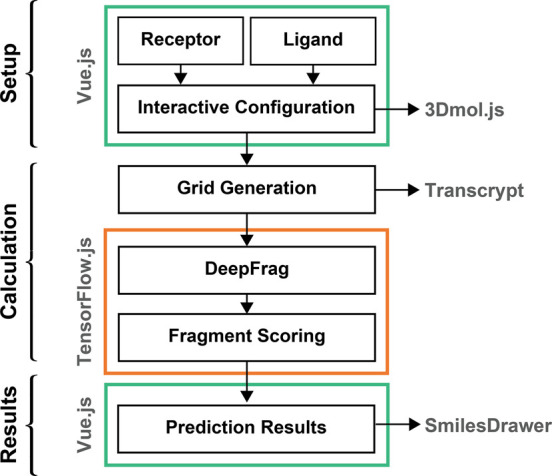
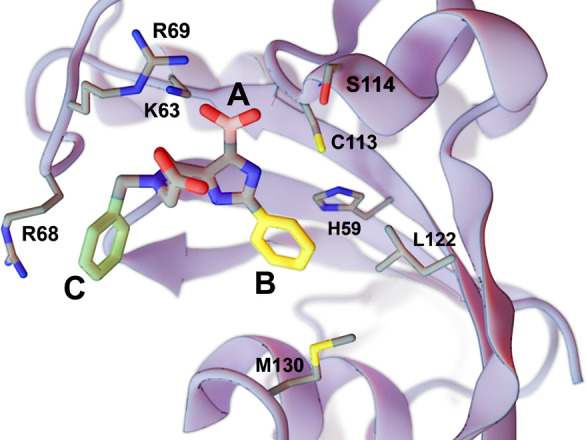
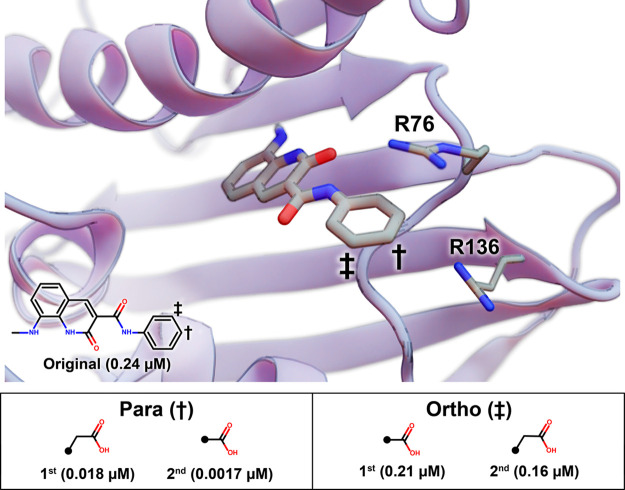
Lead optimization, a critical step in early stage drug discovery, involves making chemical modifications to a small-molecule ligand to improve properties such as binding affinity. We recently developed DeepFrag, a deep-learning model capable of recommending such modifications. Though a powerful hypothesis-generating tool, DeepFrag is currently implemented in Python and so requires a certain degree of computational expertise. To encourage broader adoption, we have created the DeepFrag browser app, which provides a user-friendly graphical user interface that runs the DeepFrag model in users' web browsers. The browser app does not require users to upload their molecular structures to a third-party server, nor does it require the separate installation of any third-party software. We are hopeful that the app will be a useful tool for both researchers and students. It can be accessed free of charge, without registration, at http://durrantlab.com/deepfrag. The source code is also available at http://git.durrantlab.com/jdurrant/deepfrag-app, released under the terms of the open-source Apache License, Version 2.0.
先导优化是药物发现早期的关键步骤,涉及对小分子配体进行化学修饰,以改善结合亲和力等性质。我们最近开发了 DeepFrag,这是一种能够推荐此类修饰的深度学习模型。尽管 DeepFrag 是一种强大的假设生成工具,但它目前是用 Python 实现的,因此需要一定程度的计算专业知识。为了鼓励更广泛的采用,我们创建了 DeepFrag 浏览器应用程序,该应用程序提供了一个用户友好的图形用户界面,可在用户的网络浏览器中运行 DeepFrag 模型。浏览器应用程序不需要用户将其分子结构上传到第三方服务器,也不需要单独安装任何第三方软件。我们希望该应用程序将成为研究人员和学生的有用工具。它可以免费访问,无需注册,网址为 http://durrantlab.com/deepfrag。源代码也可在 http://git.durrantlab.com/jdurrant/deepfrag-app 获得,根据开源 Apache License,Version 2.0 发布。