Unidad de Vinculación Científica de la Facultad de Medicina UNAM-INMEGEN, Instituto Nacional de Medicina Genómica, Periferico Sur 4809, Arenal Tepepan, Tlalpan, 14610, Mexico City, Mexico.
Programa de Doctorado en ICES, Facultad de Química, UNAM, Mexico City, Mexico.
Mol Med. 2021 May 24;27(1):50. doi: 10.1186/s10020-021-00311-5.
To evaluate the taxonomic composition of the gut microbiome in gout patients with and without tophi formation, and predict bacterial functions that might have an impact on urate metabolism.
Hypervariable V3-V4 regions of the bacterial 16S rRNA gene from fecal samples of gout patients with and without tophi (n = 33 and n = 25, respectively) were sequenced and compared to fecal samples from 53 healthy controls. We explored predictive functional profiles using bioinformatics in order to identify differences in taxonomy and metabolic pathways.
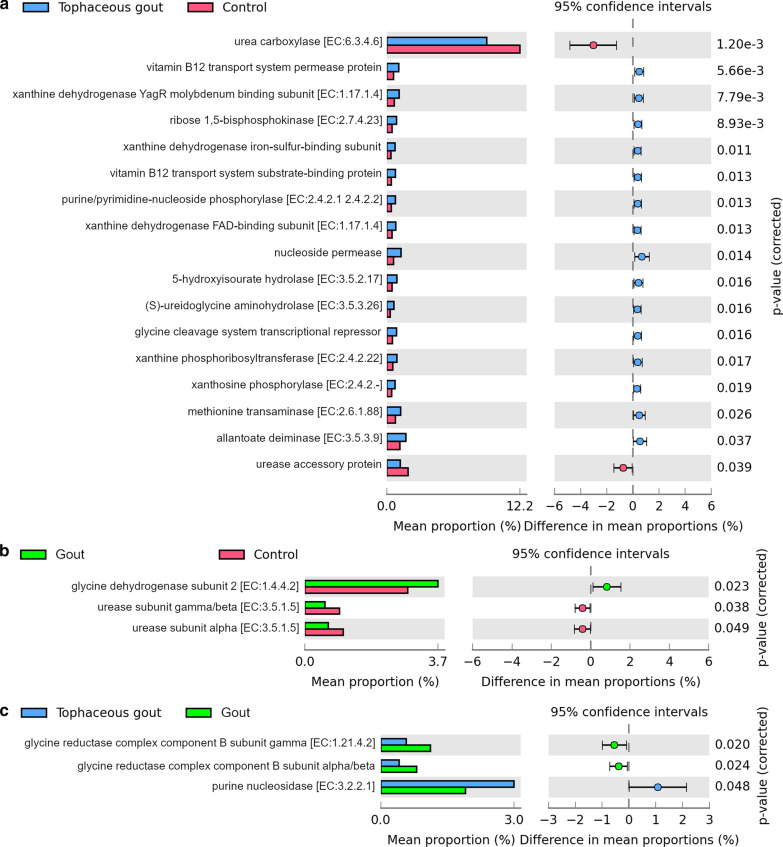
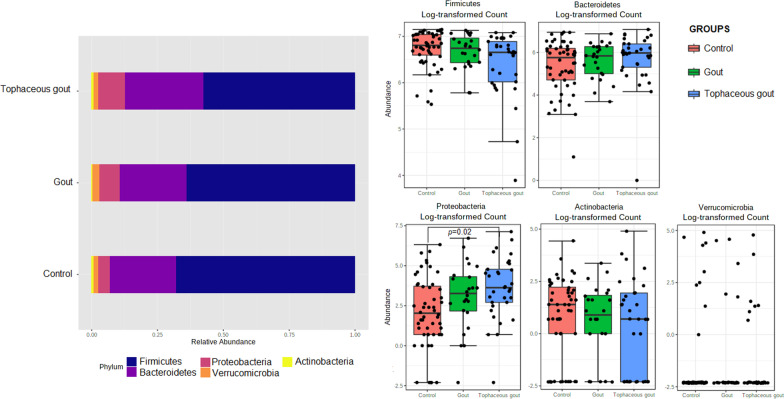
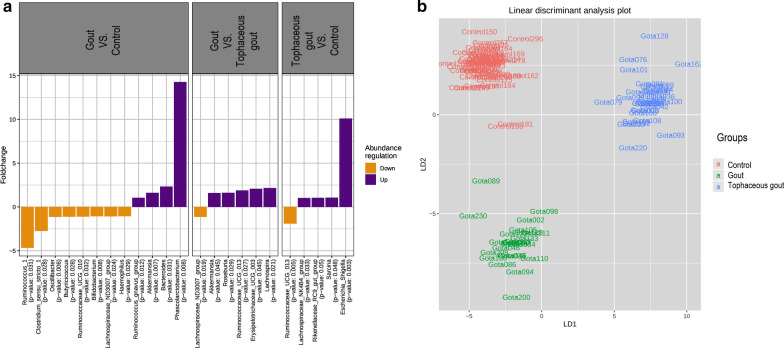
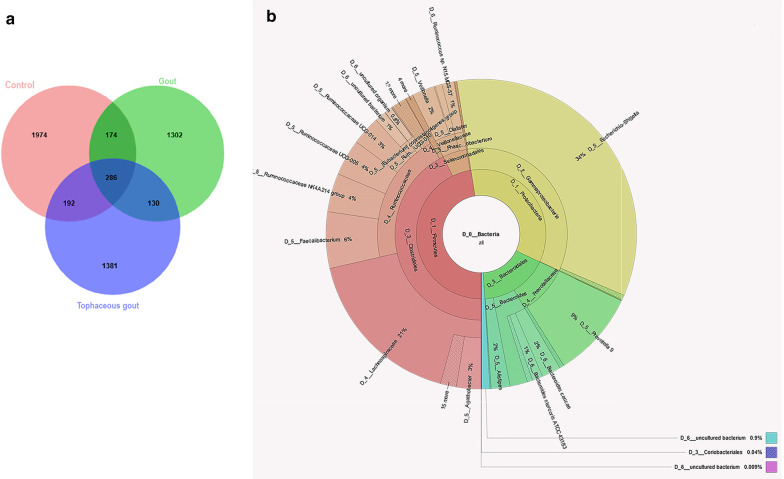
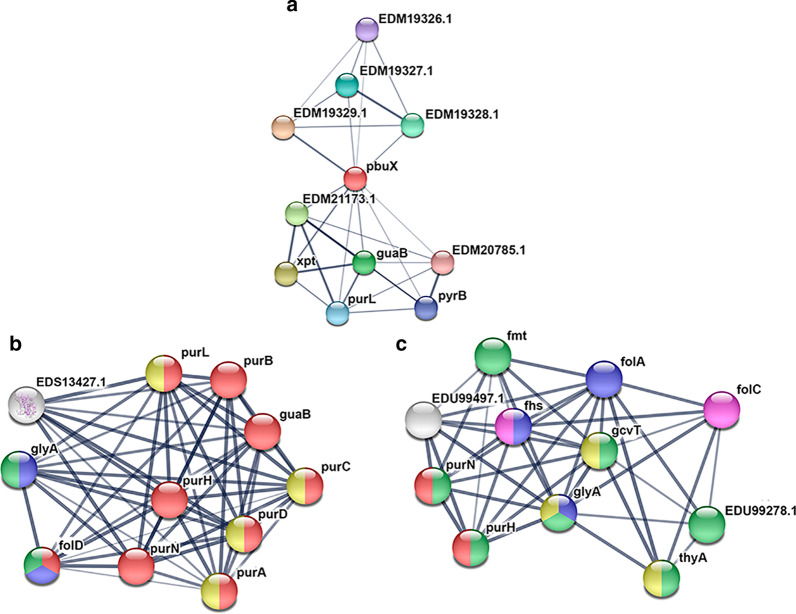
We identified a microbiome characterized by the lowest richness and a higher abundance of Phascolarctobacterium, Bacteroides, Akkermansia, and Ruminococcus_gnavus_group genera in patients with gout without tophi when compared to controls. The Proteobacteria phylum and the Escherichia-Shigella genus were more abundant in patients with tophaceous gout than in controls. Fold change analysis detected nine genera enriched in healthy controls compared to gout groups (Bifidobacterium, Butyricicoccus, Oscillobacter, Ruminococcaceae_UCG_010, Lachnospiraceae_ND2007_group, Haemophilus, Ruminococcus_1, Clostridium_sensu_stricto_1, and Ruminococcaceae_UGC_013). We found that the core microbiota of both gout groups shared Bacteroides caccae, Bacteroides stercoris ATCC 43183, and Bacteroides coprocola DSM 17136. These bacteria might perform functions linked to one-carbon metabolism, nucleotide binding, amino acid biosynthesis, and purine biosynthesis. Finally, we observed differences in key bacterial enzymes involved in urate synthesis, degradation, and elimination.
Our findings revealed that taxonomic variations in the gut microbiome of gout patients with and without tophi might have a functional impact on urate metabolism.
评估有和无痛风石形成的痛风患者肠道微生物组的分类组成,并预测可能影响尿酸代谢的细菌功能。
对有和无痛风石的痛风患者(分别为 n=33 和 n=25)的粪便样本中细菌 16S rRNA 基因的高变区 V3-V4 进行测序,并与 53 名健康对照者的粪便样本进行比较。我们使用生物信息学方法探索预测功能谱,以确定分类和代谢途径的差异。
与对照组相比,我们发现无痛风石的痛风患者的微生物组具有最低的丰富度和更高的 Phascolarctobacterium、Bacteroides、Akkermansia 和 Ruminococcus_gnavus_group 属的丰度。厚壁菌门和 Escherichia-Shigella 属在有痛风石的痛风患者中比在对照组中更为丰富。与痛风组相比,折叠变化分析检测到 9 个属在健康对照组中更为丰富(双歧杆菌、Butyricicoccus、Oscillobacter、Ruminococcaceae_UCG_010、Lachnospiraceae_ND2007_group、嗜血杆菌、Ruminococcus_1、Clostridium_sensu_stricto_1 和 Ruminococcaceae_UGC_013)。我们发现,两组痛风患者的核心微生物群都共享 Bacteroides caccae、Bacteroides stercoris ATCC 43183 和 Bacteroides coprocola DSM 17136。这些细菌可能执行与一碳代谢、核苷酸结合、氨基酸生物合成和嘌呤生物合成相关的功能。最后,我们观察到尿酸合成、降解和消除过程中关键细菌酶的差异。
我们的发现表明,有和无痛风石的痛风患者肠道微生物组的分类变化可能对尿酸代谢有功能影响。