Reis Fernanda Salles, Gomes Débora Cristiane, Arantes Henrique Pierotti, Lazaretti-Castro Marise
Departamento de Medicina, Disciplina de Endocrinologia, Universidade Federal de São Paulo (Unifesp), São Paulo, SP, Brasil.
Departamento de Medicina, Serviço de Endocrinologia Pediátrica, Universidade Federal de Uberlândia, Minas Gerais, MG, Brasil.
Arch Endocrinol Metab. 2021 May 18;64(5):623-629. doi: 10.20945/2359-3997000000222.
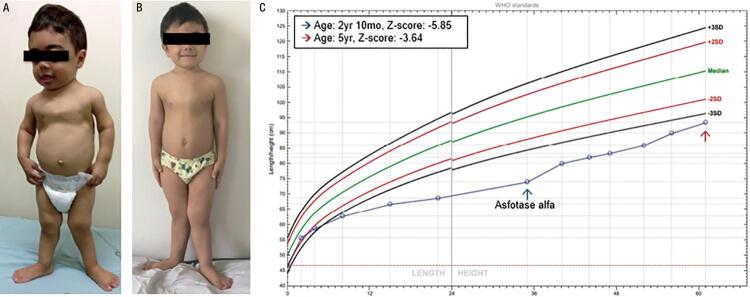
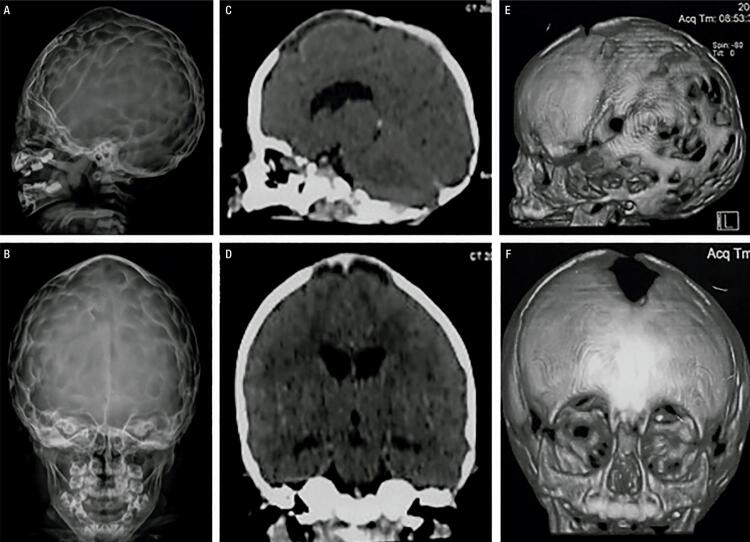
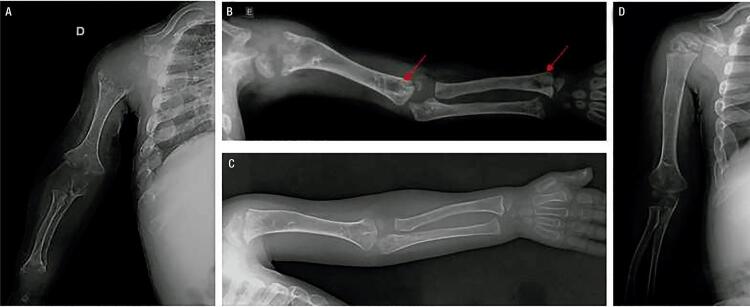
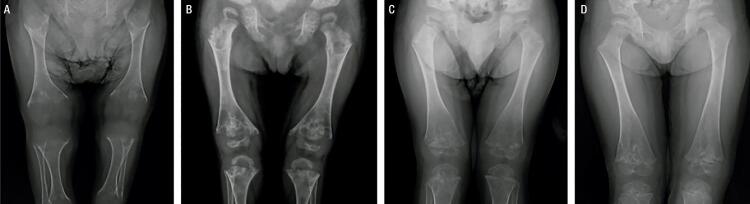
Hypophosphatasia (HPP) is a rare disease with a high mortality rate in its severe forms. It is caused by mutations within the gene encoding the tissue-nonspecific alkaline phosphatase (TNSALP), an enzyme responsible for bone mineralization. In 2015, the Food and Drug Administration approved the use of asfotase alfa, the first medication showing benefit in the treatment of HPP. We describe a case with a 2-year follow-up of the first Brazilian child treated with asfotase alfa. A 5-year-old boy, born to consanguineous parents, was diagnosed with HPP at the age of 20 months. During prenatal ultrasonography, polyhydramnios and shortening of long bones were detected. After birth, he presented delayed motor development, repeated respiratory infections, and bone deformities. At the age of 2 years and 8 months, he started walking and had already lost his primary teeth. He had reduced levels of alkaline phosphatase (ALP), elevated levels of pyridoxal 5'-phosphate (PLP), and a p.Ala33Val (c.98C>T) missense mutation in homozygosis in the gene. His parents and sister also had reduced ALP levels, high PLP levels, and the same mutation in heterozygosis. His father and sister were healthy, and his mother was diagnosed with rickets in childhood, which resulted in short physical stature and lower limb deformities. The patient was started on asfotase alfa at the age of 2 years and 10 months. After 2 years of treatment, he improved his motor skills, had no further episodes of severe respiratory infection, and showed improved radiological findings of rickets, without any severe side effect.
低磷性骨软化症(HPP)是一种罕见疾病,严重形式下死亡率很高。它由编码组织非特异性碱性磷酸酶(TNSALP)的基因突变引起,TNSALP是一种负责骨矿化的酶。2015年,美国食品药品监督管理局批准使用阿法骨化醇,这是第一种显示出对HPP治疗有益的药物。我们描述了首例接受阿法骨化醇治疗的巴西儿童,并对其进行了为期2年的随访。一名5岁男孩,其父母为近亲结婚,在20个月大时被诊断为HPP。产前超声检查发现羊水过多和长骨缩短。出生后,他出现运动发育迟缓、反复呼吸道感染和骨骼畸形。在2岁8个月时,他开始走路,乳牙已经脱落。他的碱性磷酸酶(ALP)水平降低,5'-磷酸吡哆醛(PLP)水平升高,基因中存在纯合子p.Ala33Val(c.98C>T)错义突变。他的父母和姐姐也有ALP水平降低、PLP水平升高以及杂合子形式的相同突变。他的父亲和姐姐身体健康,他的母亲在童年时被诊断为佝偻病,导致身材矮小和下肢畸形。该患者在2岁10个月时开始使用阿法骨化醇治疗。经过2年治疗,他的运动技能有所改善,没有再出现严重呼吸道感染发作,佝偻病的影像学表现也有所改善,且没有任何严重副作用。