Mathuber Marlene, Gutmann Michael, La Franca Mery, Vician Petra, Laemmerer Anna, Moser Patrick, Keppler Bernhard K, Berger Walter, Kowol Christian R
Institute of Inorganic Chemistry, Faculty of Chemistry, University of Vienna Waehringer Straße 42 1090 Vienna Austria.
Institute of Cancer Research and Comprehensive Cancer Center, Medical University of Vienna Borschkegasse 8A 1090 Vienna Austria.
Inorg Chem Front. 2021 Mar 30;8(10):2468-2485. doi: 10.1039/d1qi00211b.
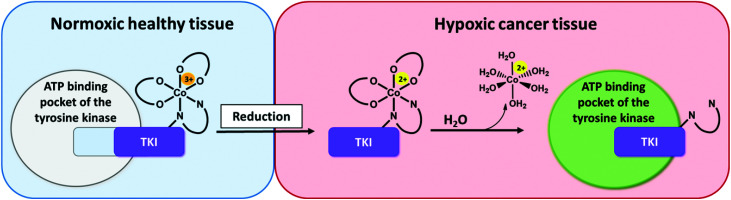
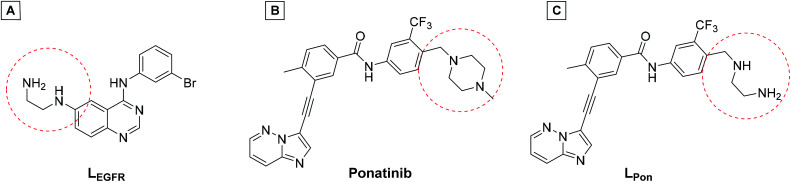
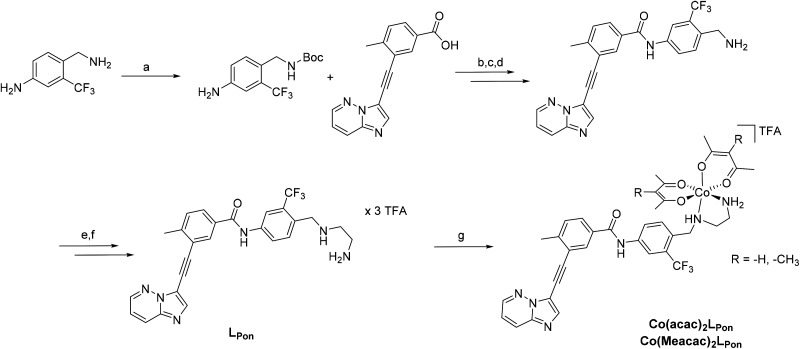
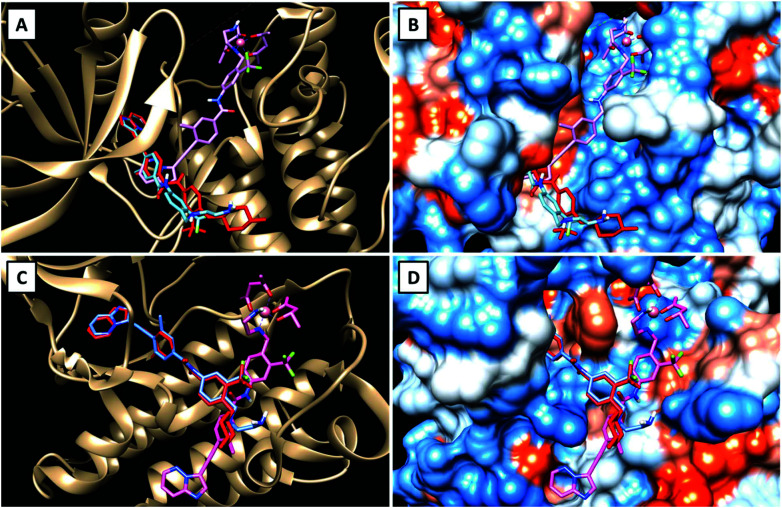
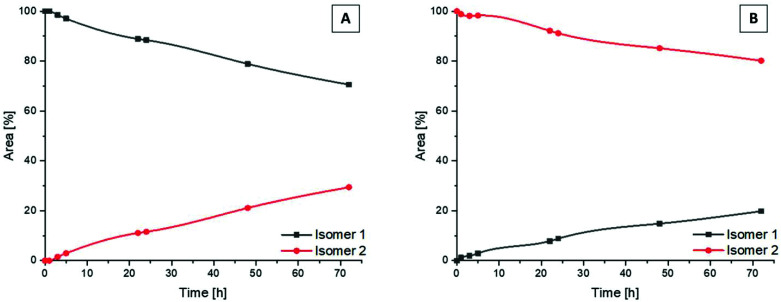
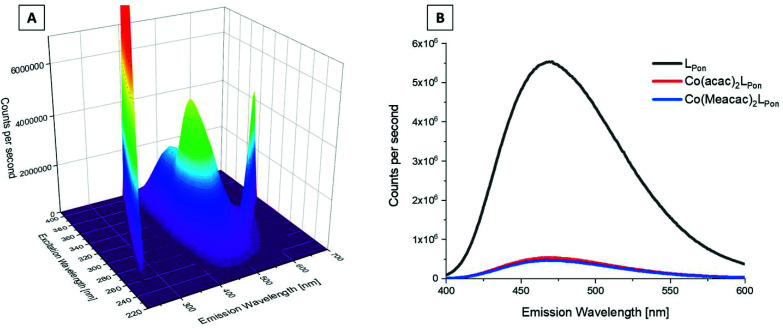
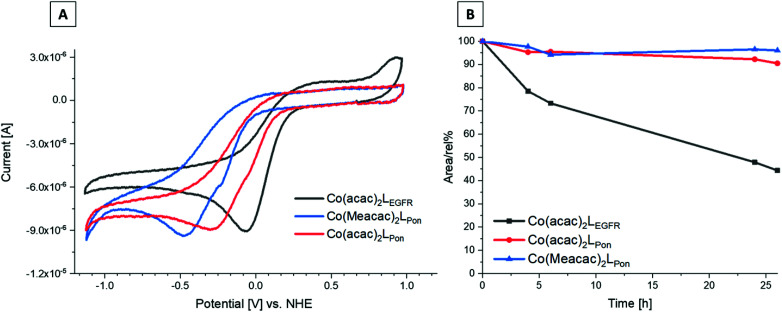
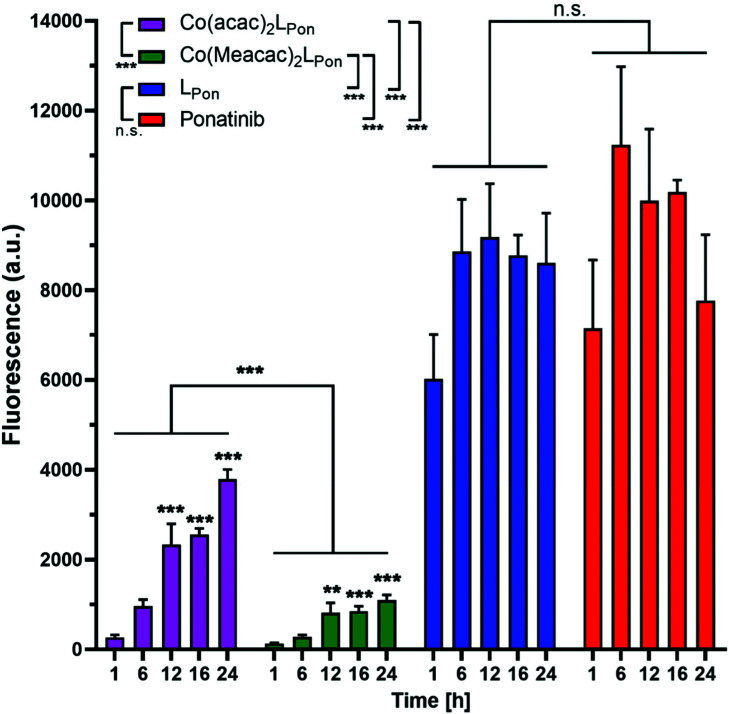
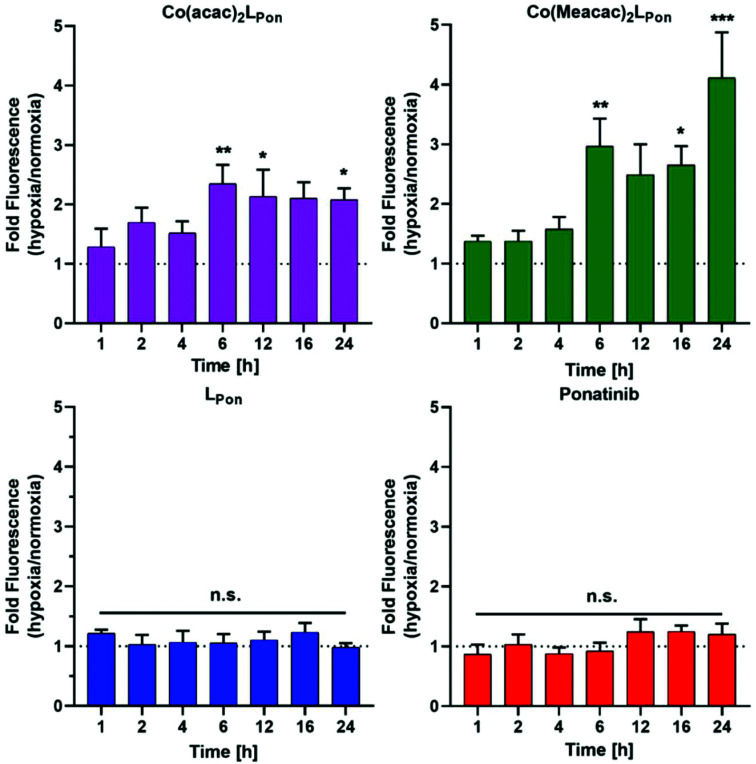
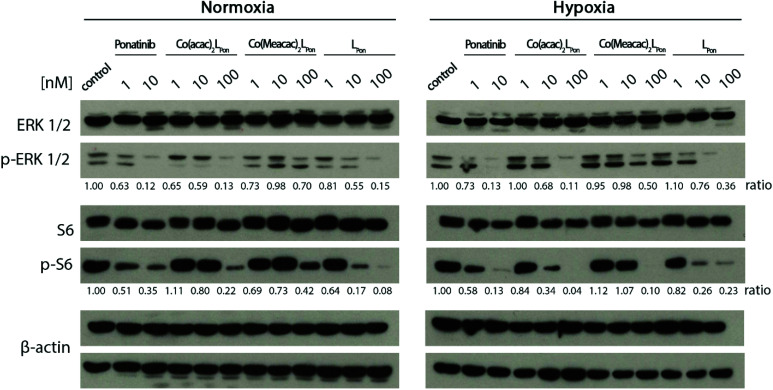
Receptor tyrosine kinase inhibitors have become a central part of modern targeted cancer therapy. However, their curative potential is distinctly limited by both rapid resistance development and severe adverse effects. Consequently, tumor-specific drug activation based on prodrug designs, exploiting tumor-specific properties such as hypoxic oxygen conditions, is a feasible strategy to widen the therapeutic window. After proof-of-principal molecular docking studies, we have synthesized two cobalt(iii) complexes using a derivative of the clinically approved Abelson (ABL) kinase and fibroblast growth factor receptor (FGFR) inhibitor ponatinib. Acetylacetone (acac) or methylacetylacetone (Meacac) have been used as ancillary ligands to modulate the reduction potential. The ponatinib derivative, characterized by an ethylenediamine moiety instead of the piperazine ring, exhibited comparable cell-free target kinase inhibition potency. Hypoxia-dependent release of the ligand from the cobalt(iii) complexes was proven by changed fluorescence properties, enhanced downstream signaling inhibition and increased anticancer activity in BCR-ABL- and FGFR-driven cancer models. Respective tumor-inhibiting effects in the BCR-ABL-driven K-562 leukemia model were restricted to the cobalt(iii) complex with the higher reduction potential and confirmed in a FGFR-driven urothelial carcinoma xenograft model. Summarizing, we here present for the first time hypoxia-activatable prodrugs of the clinically approved tyrosine kinase inhibitor ponatinib and a correlation of the activity with their reduction potential.
受体酪氨酸激酶抑制剂已成为现代靶向癌症治疗的核心组成部分。然而,它们的治疗潜力明显受到快速产生耐药性和严重不良反应的限制。因此,基于前药设计利用肿瘤特异性特性(如缺氧条件)进行肿瘤特异性药物激活是扩大治疗窗口的可行策略。在进行了原理验证分子对接研究后,我们使用临床批准的阿贝尔森(ABL)激酶和成纤维细胞生长因子受体(FGFR)抑制剂波纳替尼的衍生物合成了两种钴(III)配合物。乙酰丙酮(acac)或甲基乙酰丙酮(Meacac)已被用作辅助配体来调节还原电位。以乙二胺部分取代哌嗪环为特征的波纳替尼衍生物表现出相当的无细胞靶激酶抑制效力。通过荧光性质的改变、增强的下游信号抑制以及在BCR-ABL和FGFR驱动的癌症模型中抗癌活性的增加,证明了配体从钴(III)配合物中缺氧依赖性释放。在BCR-ABL驱动的K-562白血病模型中,相应的肿瘤抑制作用仅限于具有较高还原电位的钴(III)配合物,并在FGFR驱动的尿路上皮癌异种移植模型中得到证实。总之,我们首次展示了临床批准的酪氨酸激酶抑制剂波纳替尼的缺氧可激活前药及其活性与还原电位的相关性。