European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Hinxton, UK.
Department of Public Health Sciences, Medical University of South Carolina, Charleston, SC, USA.
BMC Bioinformatics. 2021 May 28;22(1):285. doi: 10.1186/s12859-021-04183-8.
Many important applications in bioinformatics, including sequence alignment and protein family profiling, employ sequence weighting schemes to mitigate the effects of non-independence of homologous sequences and under- or over-representation of certain taxa in a dataset. These schemes aim to assign high weights to sequences that are 'novel' compared to the others in the same dataset, and low weights to sequences that are over-represented.
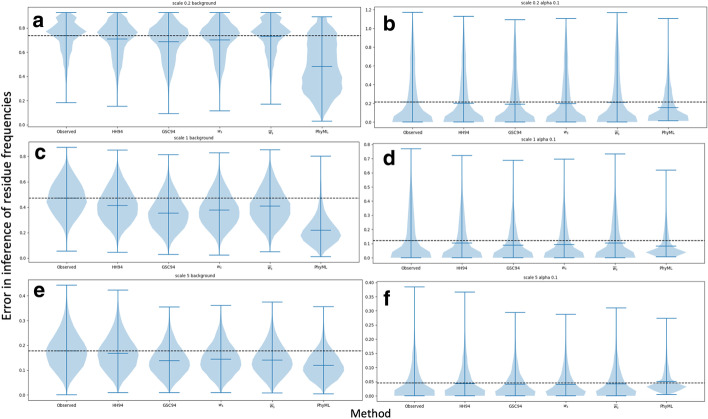
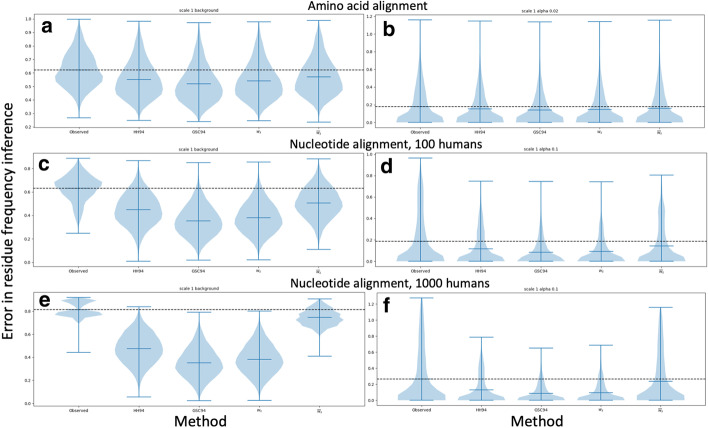
We formalise this principle by rigorously defining the evolutionary 'novelty' of a sequence within an alignment. This results in new sequence weights that we call 'phylogenetic novelty scores'. These scores have various desirable properties, and we showcase their use by considering, as an example application, the inference of character frequencies at an alignment column-important, for example, in protein family profiling. We give computationally efficient algorithms for calculating our scores and, using simulations, show that they are versatile and can improve the accuracy of character frequency estimation compared to existing sequence weighting schemes.
Our phylogenetic novelty scores can be useful when an evolutionarily meaningful system for adjusting for uneven taxon sampling is desired. They have numerous possible applications, including estimation of evolutionary conservation scores and sequence logos, identification of targets in conservation biology, and improving and measuring sequence alignment accuracy.
生物信息学中的许多重要应用,包括序列比对和蛋白质家族分析,都采用序列加权方案来减轻同源序列的非独立性以及数据集中某些分类单元的过表达或欠表达的影响。这些方案旨在为与同一数据集中的其他序列相比具有“新颖性”的序列分配高权重,而为过表达的序列分配低权重。
我们通过严格定义比对中序列的进化“新颖性”来形式化这一原则。这导致了我们称之为“系统发育新颖性得分”的新序列权重。这些得分具有各种理想的特性,我们通过考虑一个示例应用来展示它们的用途,例如在对齐列字符频率推断中的应用,这对于蛋白质家族分析非常重要。我们给出了计算我们得分的计算效率算法,并通过模拟表明,与现有序列加权方案相比,它们具有多功能性,可以提高字符频率估计的准确性。
当需要一个具有进化意义的系统来调整不均匀的分类单元采样时,我们的系统发育新颖性得分可能非常有用。它们有许多可能的应用,包括估计进化保守性得分和序列标志,识别保护生物学中的目标,以及改进和衡量序列比对的准确性。