Department of Infectious Diseases, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China.
Int J Immunopathol Pharmacol. 2021 Jan-Dec;35:20587384211018389. doi: 10.1177/20587384211018389.
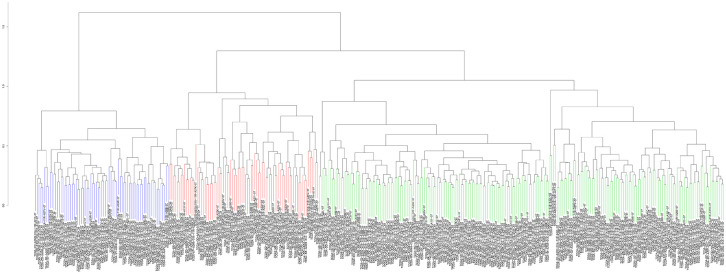
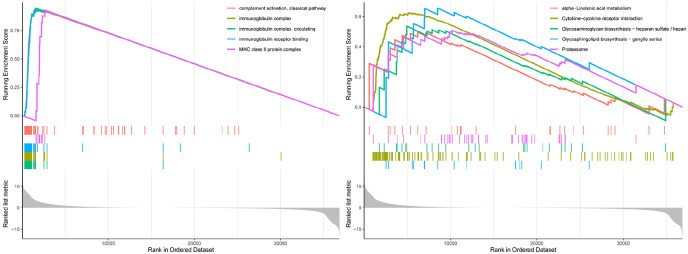
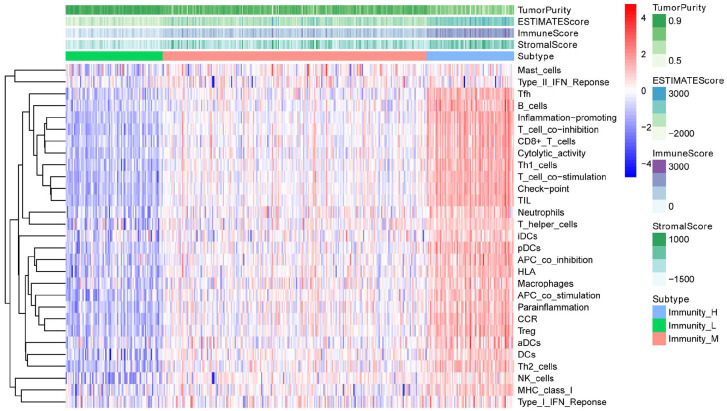
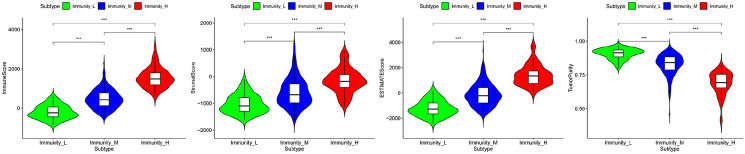
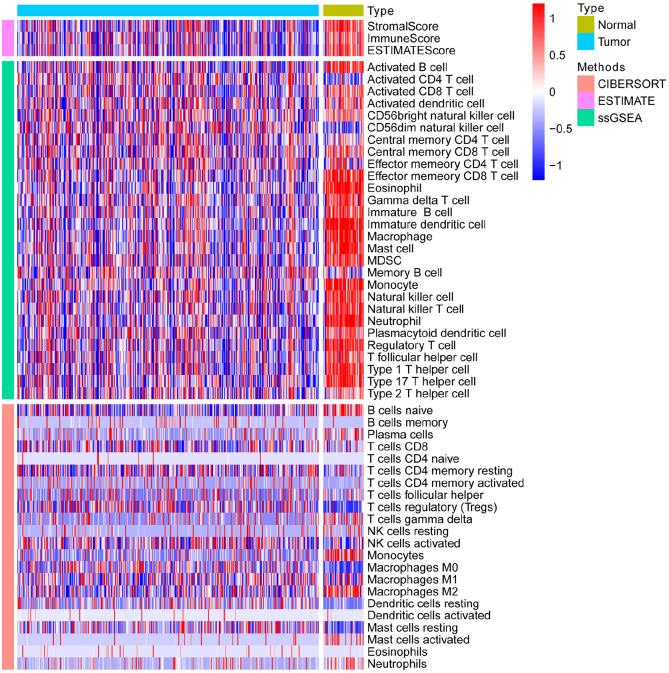
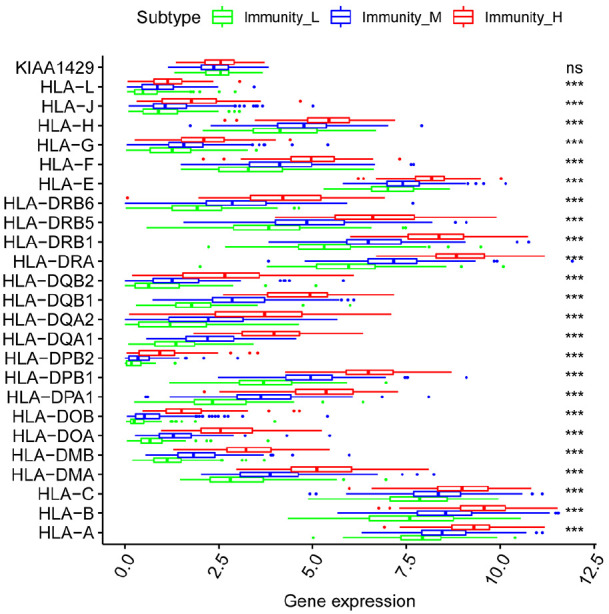
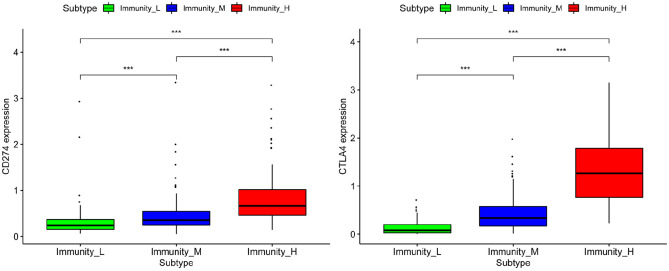
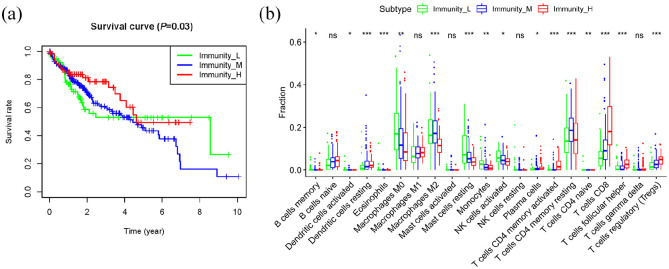
Hepatocellular carcinoma (HCC) is a high mortality malignancy and the second leading cause of cancer-related deaths. Because the immune system plays a dual role by assisting the host barrier and tumor progression, there are complex interactions with considerable prognostic significance. Herein, we performed single-sample gene set enrichment (ssGSEA) to explore the tumor microenvironment (TME) and quantify the tumor-infiltrating immune cell (TIIC) subgroups of immune responses based on the HCC cohort of The Cancer Genome Atlas (TCGA) database. We evaluate molecular subpopulations, survival, function, and expression differential associations, as well as reveal potential targets, and biomarkers for immunotherapy. We combined the TME score and the 29 immune cell types in the low, medium, and high immunity groups. The stromal score, immune score, and ESTIMATE score were positively correlated with immune activity but negatively correlated with the tumor purity. There were 23 human leukocyte antigen (HLA)-related genes that were significantly different. However, KIAA1429 was not significant among the different immunity groups. Besides, programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) expression increased with the increase of immune activity. This may provide valuable information for HCC immunotherapy. We also found that there was no significant difference in naïve B cells, macrophages M1, activated mast cells, resting natural killer (NK) cells, and T cells gamma delta among the different immunity groups. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that the differential proteins were mainly enriched in alpha-linolenic acid (ALA) metabolism, cytokine-cytokine receptor interaction, glycosaminoglycan biosynthesis-heparan sulfate/heparin, glycosphingolipid biosynthesis-ganglio series and proteasome. Our findings provide a deeper understanding of the immune scene, uncovering remarkable immune infiltration patterns of various subtypes of HCC using ssGSEA. This study advances the understanding of immune response and provides a basis for research to enhance immunotherapy.
肝细胞癌(HCC)是一种高死亡率的恶性肿瘤,也是癌症相关死亡的第二大主要原因。由于免疫系统通过协助宿主屏障和肿瘤进展发挥双重作用,因此与具有重要预后意义的复杂相互作用有关。在此,我们基于癌症基因组图谱(TCGA)数据库的 HCC 队列进行了单样本基因集富集分析(ssGSEA),以探索肿瘤微环境(TME)并量化肿瘤浸润免疫细胞(TIIC)亚群的免疫反应。我们评估了分子亚群、生存、功能和表达差异关联,以及揭示潜在的免疫治疗靶点和生物标志物。我们将 TME 评分与低、中、高免疫组的 29 种免疫细胞类型相结合。基质评分、免疫评分和 ESTIMATE 评分与免疫活性呈正相关,与肿瘤纯度呈负相关。有 23 个人类白细胞抗原(HLA)相关基因在不同免疫组之间存在显著差异。然而,在不同免疫组之间,KIAA1429 并不显著。此外,程序性死亡配体 1(PD-L1)和细胞毒性 T 淋巴细胞相关抗原 4(CTLA-4)的表达随着免疫活性的增加而增加。这可能为 HCC 免疫治疗提供有价值的信息。我们还发现,在不同免疫组之间,幼稚 B 细胞、巨噬细胞 M1、活化肥大细胞、静止自然杀伤(NK)细胞和 T 细胞γδ之间没有显著差异。京都基因与基因组百科全书(KEGG)通路分析显示,差异蛋白主要富集在α-亚麻酸(ALA)代谢、细胞因子-细胞因子受体相互作用、糖胺聚糖生物合成-硫酸乙酰肝素/肝素、糖脂生物合成-神经节苷脂系列和蛋白酶体。我们的研究结果提供了对免疫场景的更深入了解,通过 ssGSEA 揭示了各种 HCC 亚型的显著免疫浸润模式。这项研究增进了对免疫反应的理解,并为增强免疫治疗的研究提供了基础。