Department of Chemistry, School of Basic Science, Rani Channamma University, Belagavi 591156, Karnataka, India.
Institute for Stem Cell Science and Regenerative Medicine, NCBS, TIFR, GKVK-Campus Bellary road, Bengaluru 560065, Karnataka, India.
Molecules. 2021 May 6;26(9):2722. doi: 10.3390/molecules26092722.
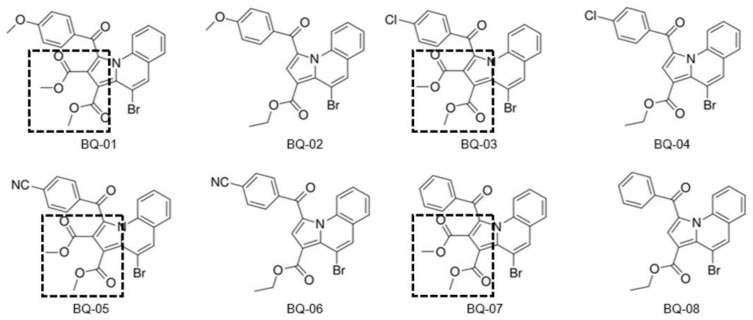
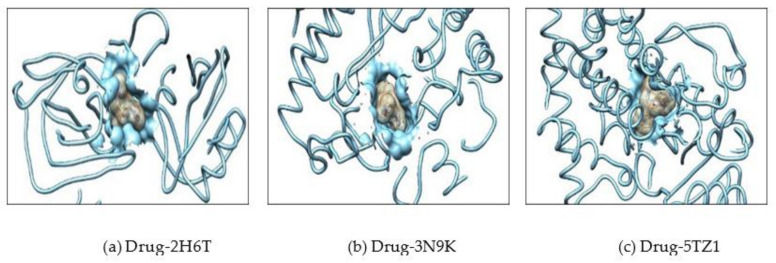
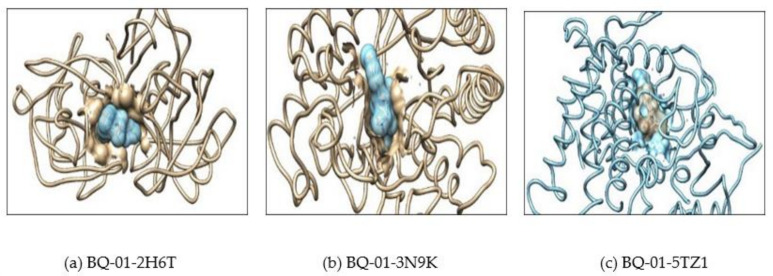
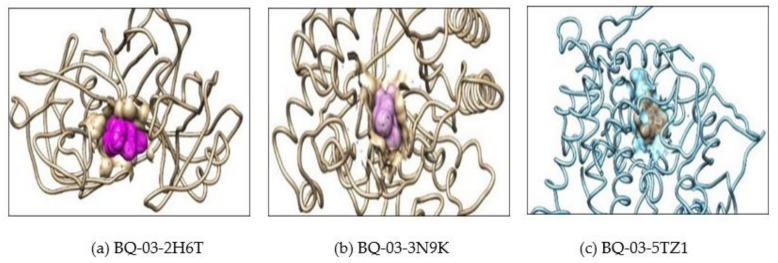
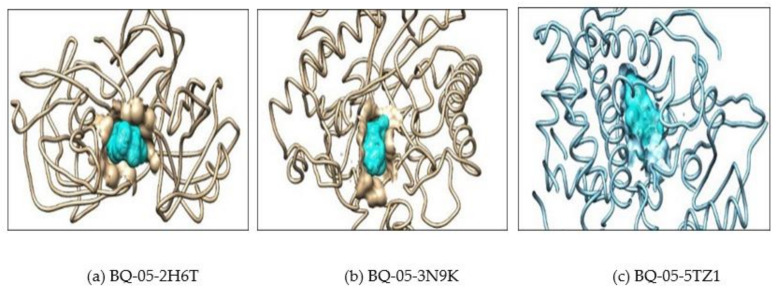
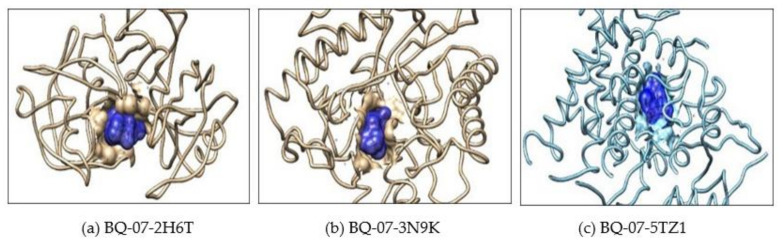
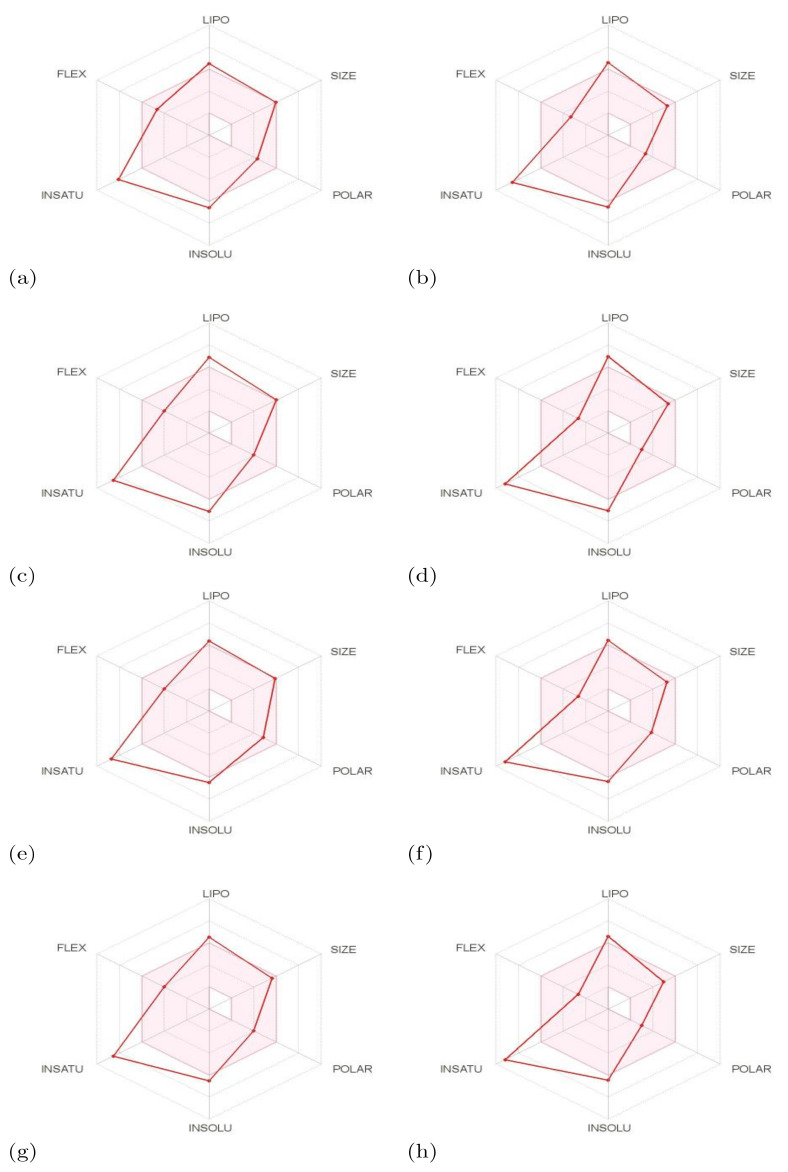
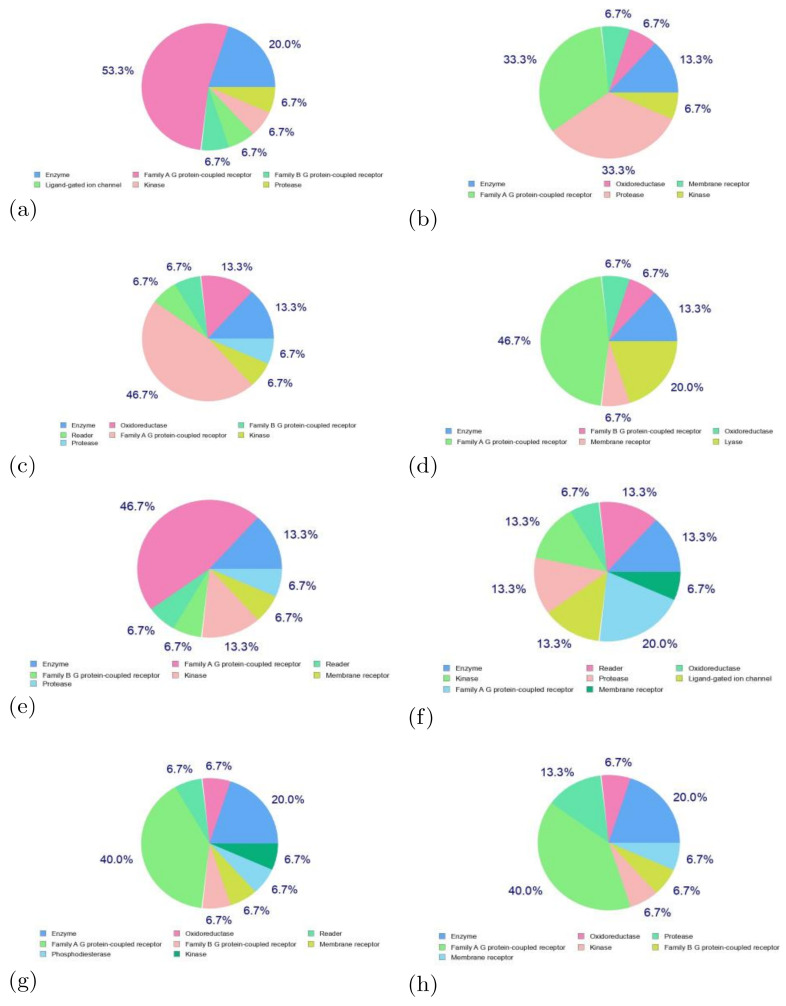
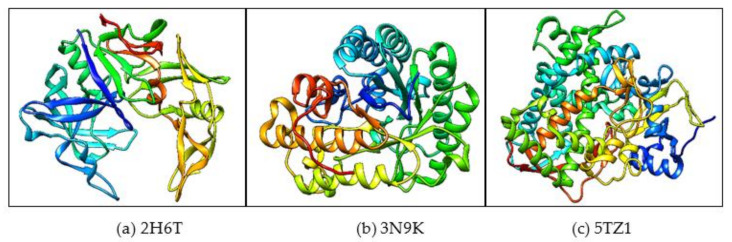
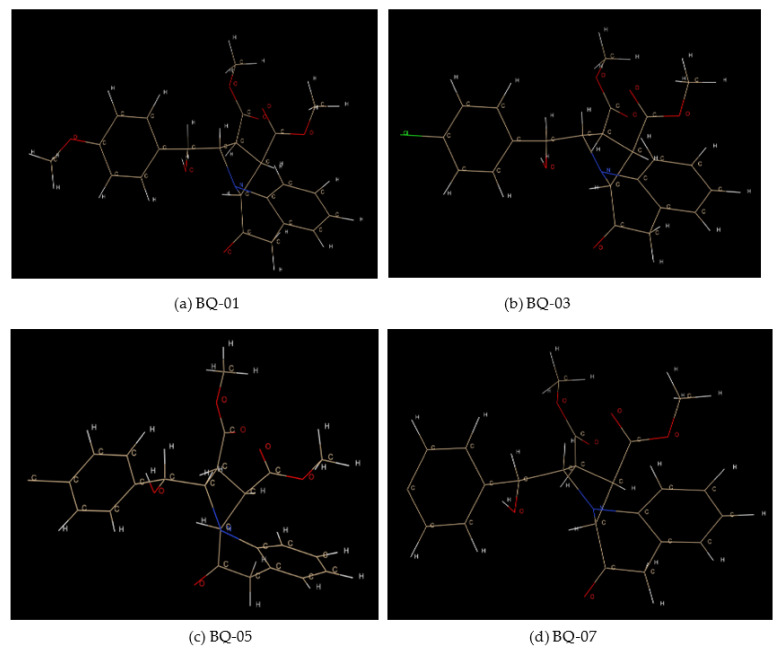
, an opportunistic fungal pathogen, frequently colonizes immune-compromised patients and causes mild to severe systemic reactions. Only few antifungal drugs are currently in use for therapeutic treatment. However, evolution of a drug-resistant fungal pathogen is of major concern in the treatment of patients, hence the clinical need for novel drug design and development. In this study, in vitro of novel putative pyrrolo[1,2-a]quinoline derivatives as the lead drug targets and in silico prediction of the binding potential of these lead molecules against pathogenic proteins, such as secreted aspartic protease 3 (SAP3; 2H6T), surface protein β-glucanase (3N9K) and sterol 14-alpha demethylase (5TZ1), were carried out by molecular docking analyses. Further, biological activity-based QSAR and theoretical pharmacokinetic analysis were analyzed. Here, in vitro screening of novel analogue derivatives as drug targets against showed inhibitory potential in the concentration of 0.4 µg for BQ-06, 07 and 08, 0.8 µg for BQ-01, 03, and 05, 1.6 µg for BQ-04 and 12.5 µg for BQ-02 in comparison to the standard antifungal drug fluconazole in the concentration of 30 µg. Further, in silico analysis of BQ-01, 03, 05 and 07 analogues docked on chimeric 2H6T, 3N9K and 5TZ1 revealed that these analogues show potential binding affinity, which is different from the therapeutic antifungal drug fluconazole. In addition, these molecules possess good drug-like properties based on the determination of conceptual Density Functional Theory (DFT)-based descriptors, QSAR and pharmacokinetics. Thus, the study offers significant insight into employing pyrrolo[1,2-a]quinoline analogues as novel antifungal agents against that warrants further investigation.
新型吡咯并[1,2-a]喹啉类衍生物作为潜在的抗真菌药物靶点,通过分子对接分析对其与致病蛋白(如分泌型天冬氨酸蛋白酶 3(SAP3;2H6T)、表面蛋白β-葡聚糖酶(3N9K)和甾醇 14-α去甲基酶(5TZ1))的结合潜力进行了计算机模拟预测。此外,还进行了基于生物活性的定量构效关系和理论药代动力学分析。在此,新型类似物衍生物作为抗真菌药物靶点对进行了体外筛选,结果表明,BQ-06、07 和 08 的抑制浓度为 0.4 µg,BQ-01、03 和 05 的抑制浓度为 0.8 µg,BQ-04 的抑制浓度为 1.6 µg,BQ-02 的抑制浓度为 12.5 µg,而标准抗真菌药物氟康唑的抑制浓度为 30 µg。此外,BQ-01、03、05 和 07 类似物与嵌合 2H6T、3N9K 和 5TZ1 的计算机模拟对接分析表明,这些类似物显示出潜在的结合亲和力,与治疗性抗真菌药物氟康唑不同。此外,这些分子基于概念性密度泛函理论(DFT)基描述符、QSAR 和药代动力学的测定,具有良好的类药性。因此,该研究为使用吡咯并[1,2-a]喹啉类似物作为新型抗真菌药物提供了重要的见解,值得进一步研究。