Pakdaman Yasaman, Berland Siren, Bustad Helene J, Erdal Sigrid, Thompson Bryony A, James Paul A, Power Kjersti N, Ellingsen Ståle, Krooni Martin, Berge Line I, Sexton Adrienne, Bindoff Laurence A, Knappskog Per M, Johansson Stefan, Aukrust Ingvild
Department of Medical Genetics, Haukeland University Hospital, 5021 Bergen, Norway.
Department of Clinical Science, University of Bergen, 5021 Bergen, Norway.
Int J Mol Sci. 2021 May 30;22(11):5870. doi: 10.3390/ijms22115870.
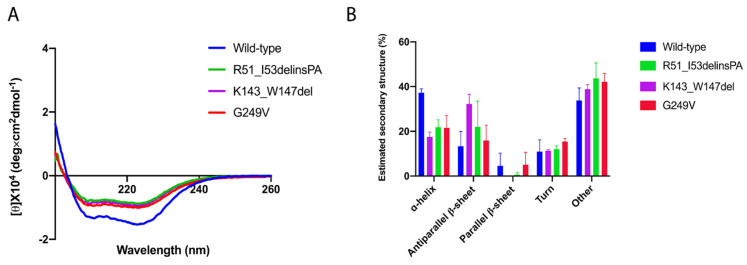
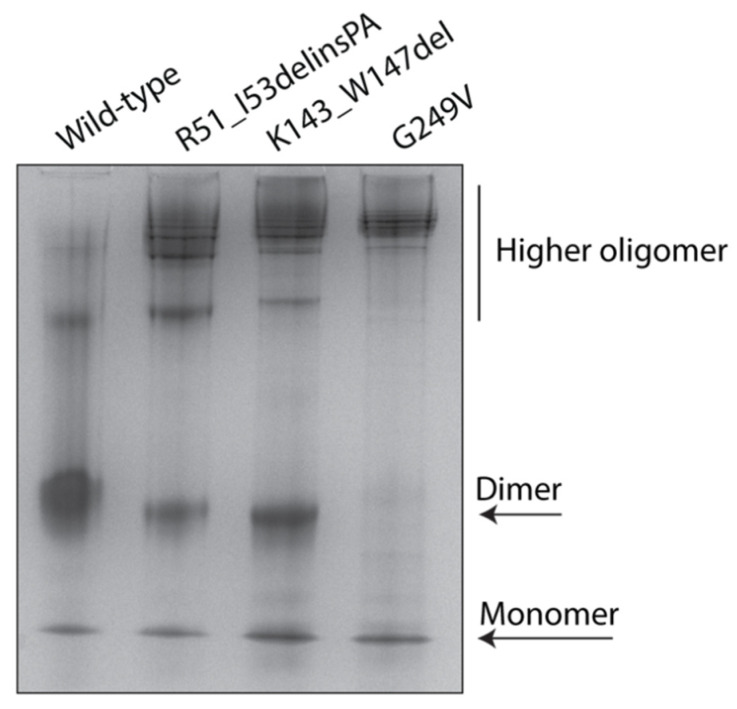
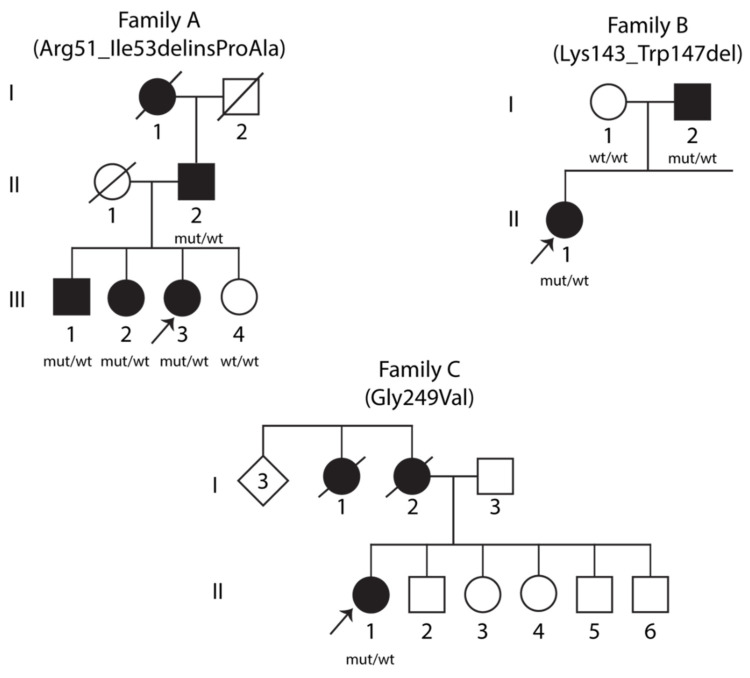
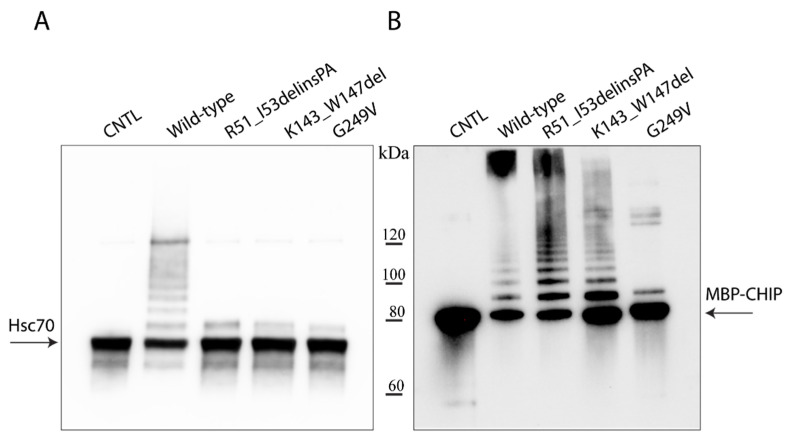
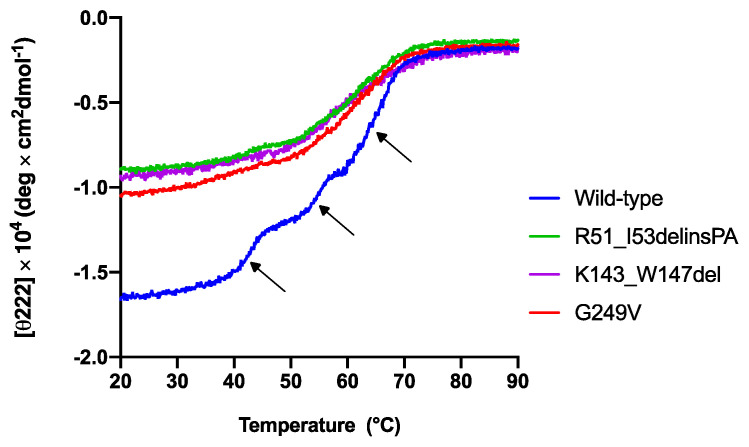
Variants in cause both autosomal recessive (SCAR16) and dominant (SCA48) spinocerebellar ataxia. Reports from 18 variants causing SCA48 show that the clinical picture includes later-onset ataxia with a cerebellar cognitive affective syndrome and varying clinical overlap with SCAR16. However, little is known about the molecular properties of dominant variants. Here, we describe three SCA48 families with novel, dominantly inherited variants (p.Arg51_Ile53delinsProAla, p.Lys143_Trp147del, and p.Gly249Val). All the patients developed symptoms from 30 years of age or later, all had cerebellar atrophy, and 4 had cognitive/psychiatric phenotypes. Investigation of the structural and functional consequences of the recombinant C-terminus of HSC70-interacting protein (CHIP) variants was performed in vitro using ubiquitin ligase activity assay, circular dichroism assay and native polyacrylamide gel electrophoresis. These studies revealed that dominantly and recessively inherited variants showed similar biochemical defects, including impaired ubiquitin ligase activity and altered oligomerization properties of the CHIP. Our findings expand the molecular understanding of SCA48 but also mean that assumptions concerning unaffected carriers of recessive variants in SCAR16 families must be re-evaluated. More investigations are needed to verify the disease status of SCAR16 heterozygotes and elucidate the molecular relationship between SCA48 and SCAR16 diseases.
基因变异可导致常染色体隐性遗传性(SCAR16)和显性遗传性(SCA48)脊髓小脑共济失调。18个导致SCA48的基因变异报告显示,临床症状包括迟发性共济失调并伴有小脑认知情感综合征,且与SCAR16存在不同程度的临床重叠。然而,对于显性基因变异的分子特性知之甚少。在此,我们描述了三个携带新型显性遗传基因变异(p.Arg51_Ile53delinsProAla、p.Lys143_Trp147del和p.Gly249Val)的SCA48家系。所有患者均在30岁及以后出现症状,均有小脑萎缩,4例有认知/精神方面的表型。利用泛素连接酶活性测定、圆二色性测定和天然聚丙烯酰胺凝胶电泳,在体外对热休克蛋白70相互作用蛋白(CHIP)变异体的重组C末端的结构和功能后果进行了研究。这些研究表明,显性和隐性遗传的基因变异表现出相似的生化缺陷,包括泛素连接酶活性受损和CHIP的寡聚化特性改变。我们的研究结果扩展了对SCA48的分子认识,但也意味着必须重新评估关于SCAR16家系中隐性基因变异未受影响携带者的假设。需要更多研究来验证SCAR16杂合子的疾病状态,并阐明SCA48与SCAR16疾病之间的分子关系。