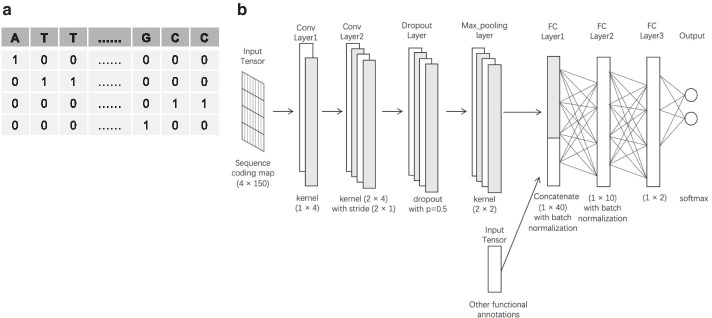
Department of Computer Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan.
Human Genome Center, the Institute of Medical Science, The University of Tokyo, 4-6-1 Shirokanedai, Minato-ku, Tokyo, 108-8639, Japan.
BMC Bioinformatics. 2021 Jun 2;22(Suppl 6):128. doi: 10.1186/s12859-021-03999-8.
Understanding the functional effects of non-coding variants is important as they are often associated with gene-expression alteration and disease development. Over the past few years, many computational tools have been developed to predict their functional impact. However, the intrinsic difficulty in dealing with the scarcity of data leads to the necessity to further improve the algorithms. In this work, we propose a novel method, employing a semi-supervised deep-learning model with pseudo labels, which takes advantage of learning from both experimentally annotated and unannotated data.
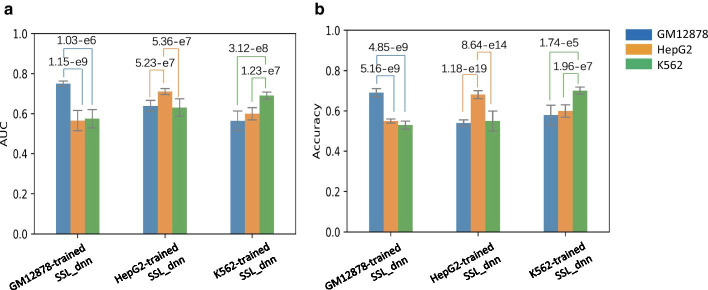
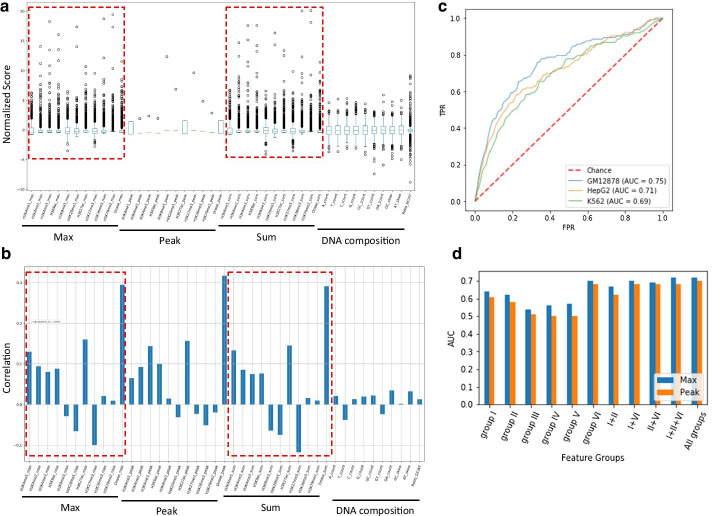
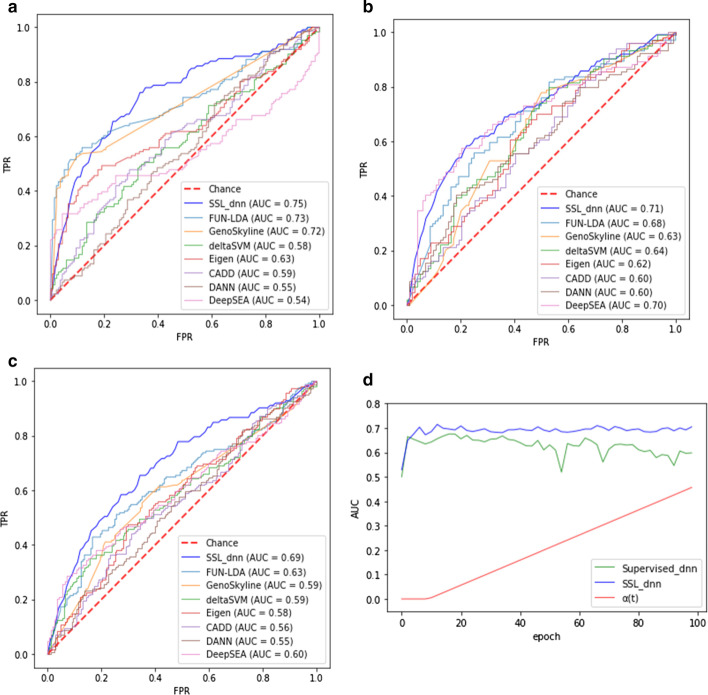
We prepared known functional non-coding variants with histone marks, DNA accessibility, and sequence context in GM12878, HepG2, and K562 cell lines. Applying our method to the dataset demonstrated its outstanding performance, compared with that of existing tools. Our results also indicated that the semi-supervised model with pseudo labels achieves higher predictive performance than the supervised model without pseudo labels. Interestingly, a model trained with the data in a certain cell line is unlikely to succeed in other cell lines, which implies the cell-type-specific nature of the non-coding variants. Remarkably, we found that DNA accessibility significantly contributes to the functional consequence of variants, which suggests the importance of open chromatin conformation prior to establishing the interaction of non-coding variants with gene regulation.
The semi-supervised deep learning model coupled with pseudo labeling has advantages in studying with limited datasets, which is not unusual in biology. Our study provides an effective approach in finding non-coding mutations potentially associated with various biological phenomena, including human diseases.
理解非编码变异的功能效应很重要,因为它们通常与基因表达改变和疾病发展有关。在过去的几年中,已经开发出许多计算工具来预测它们的功能影响。然而,处理数据稀缺性的固有困难导致需要进一步改进算法。在这项工作中,我们提出了一种新的方法,采用具有伪标签的半监督深度学习模型,利用来自实验注释和未注释数据的学习。
我们在 GM12878、HepG2 和 K562 细胞系中准备了具有组蛋白标记、DNA 可及性和序列上下文的已知功能非编码变体。将我们的方法应用于数据集表明,与现有工具相比,它具有出色的性能。我们的结果还表明,具有伪标签的半监督模型比没有伪标签的监督模型具有更高的预测性能。有趣的是,在特定细胞系中训练的模型不太可能在其他细胞系中成功,这意味着非编码变体具有细胞类型特异性。值得注意的是,我们发现 DNA 可及性对变体的功能后果有显著贡献,这表明在建立非编码变体与基因调控相互作用之前,开放染色质构象的重要性。
结合伪标签的半监督深度学习模型在处理有限数据集方面具有优势,这在生物学中并不罕见。我们的研究为寻找与各种生物学现象(包括人类疾病)相关的非编码突变提供了一种有效方法。